

ampliMethProfiler: a pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons
- PMID: 27884103
- PMCID: PMC5123276
- DOI: 10.1186/s12859-016-1380-3
ampliMethProfiler: a pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons
Abstract
Background: CpG sites in an individual molecule may exist in a binary state (methylated or unmethylated) and each individual DNA molecule, containing a certain number of CpGs, is a combination of these states defining an epihaplotype. Classic quantification based approaches to study DNA methylation are intrinsically unable to fully represent the complexity of the underlying methylation substrate. Epihaplotype based approaches, on the other hand, allow methylation profiles of cell populations to be studied at the single molecule level. For such investigations, next-generation sequencing techniques can be used, both for quantitative and for epihaplotype analysis. Currently available tools for methylation analysis lack output formats that explicitly report CpG methylation profiles at the single molecule level and that have suited statistical tools for their interpretation.
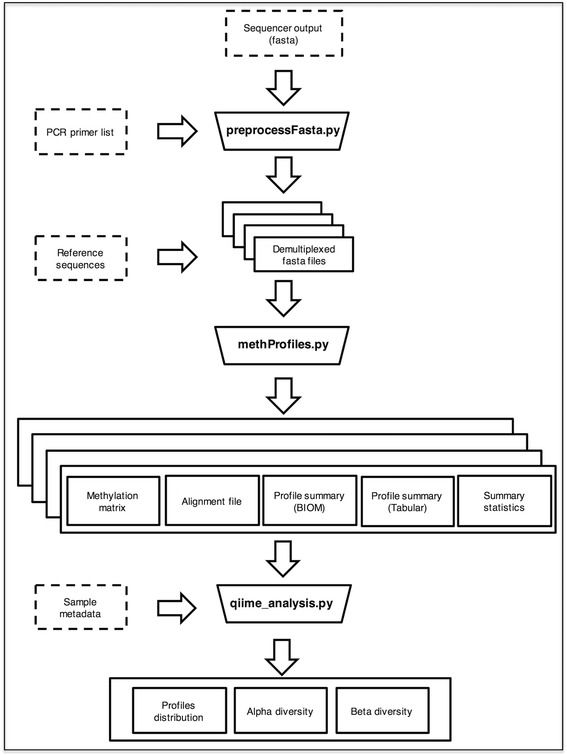
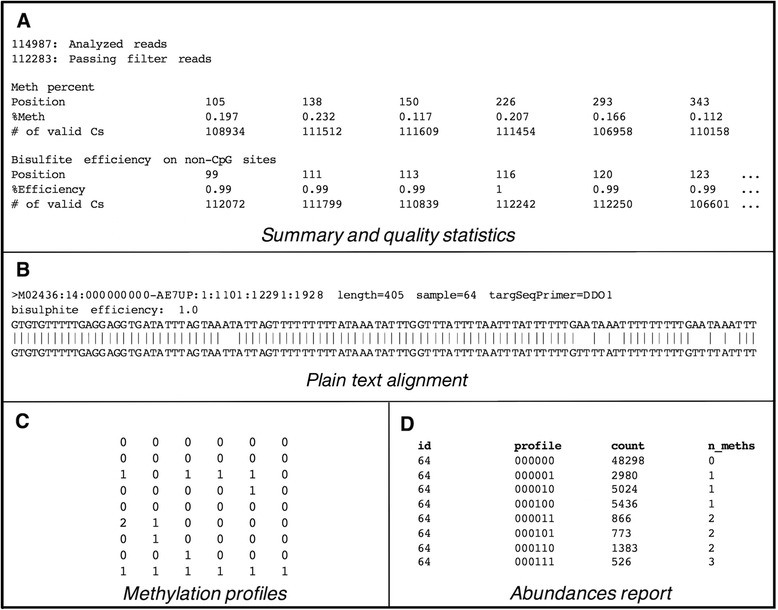
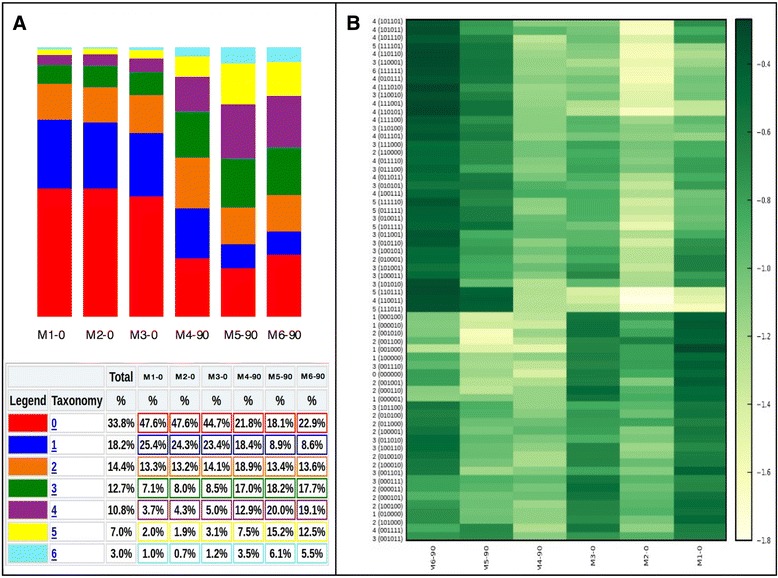
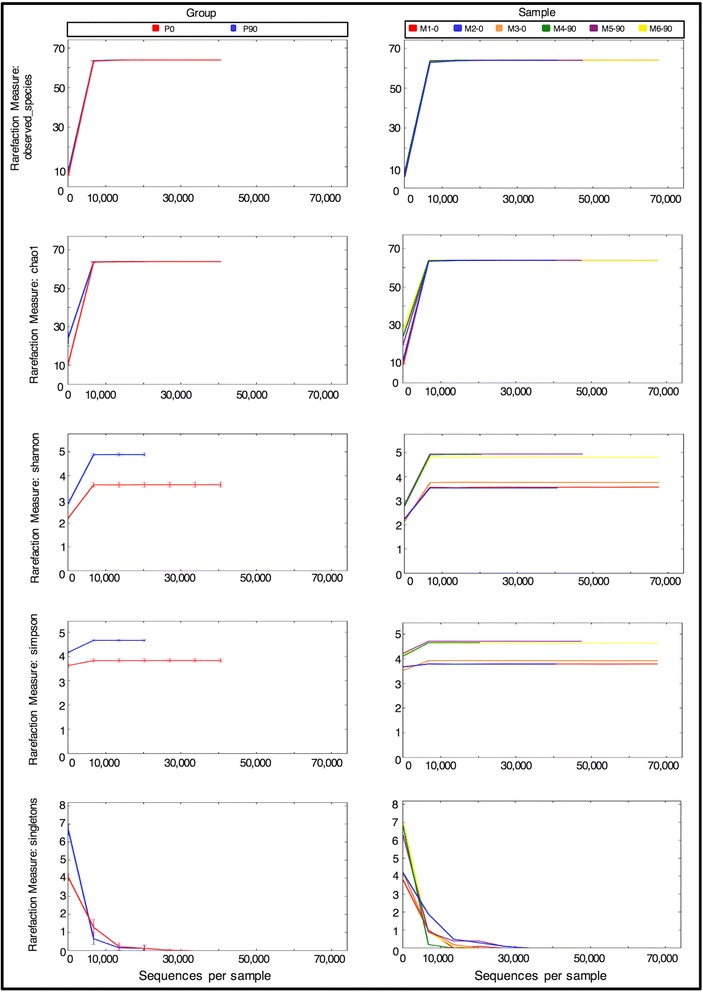
Results: Here we present ampliMethProfiler, a python-based pipeline for the extraction and statistical epihaplotype analysis of amplicons from targeted deep bisulfite sequencing of multiple DNA regions.
Conclusions: ampliMethProfiler tool provides an easy and user friendly way to extract and analyze the epihaplotype composition of reads from targeted bisulfite sequencing experiments. ampliMethProfiler is written in python language and requires a local installation of BLAST and (optionally) QIIME tools. It can be run on Linux and OS X platforms. The software is open source and freely available at http://amplimethprofiler.sourceforge.net .
Keywords: Bisulfite sequencing; DNA methylation; Epihaplotype; Epihaplotype based analysis; Methylation profiles.
Figures
Similar articles
-
DNA Methylation Profiling Using Long-Read Single Molecule Real-Time Bisulfite Sequencing (SMRT-BS).Methods Mol Biol. 2017;1654:125-134. doi: 10.1007/978-1-4939-7231-9_8. Methods Mol Biol. 2017. PMID: 28986786
-
Locus-Specific DNA Methylation Analysis by Targeted Deep Bisulfite Sequencing.Methods Mol Biol. 2018;1767:351-366. doi: 10.1007/978-1-4939-7774-1_19. Methods Mol Biol. 2018. PMID: 29524145
-
A comprehensive evaluation of alignment software for reduced representation bisulfite sequencing data.Bioinformatics. 2018 Aug 15;34(16):2715-2723. doi: 10.1093/bioinformatics/bty174. Bioinformatics. 2018. PMID: 29579198
-
Methodological aspects of whole-genome bisulfite sequencing analysis.Brief Bioinform. 2015 May;16(3):369-79. doi: 10.1093/bib/bbu016. Epub 2014 May 27. Brief Bioinform. 2015. PMID: 24867940 Review.
-
Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis.Epigenomics. 2015 Aug;7(5):813-28. doi: 10.2217/epi.15.21. Epub 2015 Sep 14. Epigenomics. 2015. PMID: 26366945 Free PMC article. Review.
Cited by
-
A novel workflow for the qualitative analysis of DNA methylation data.Comput Struct Biotechnol J. 2022 Oct 23;20:5925-5934. doi: 10.1016/j.csbj.2022.10.027. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36382198 Free PMC article.
-
The Metallophosphoesterase-Domain-Containing Protein 2 (MPPED2) Gene Acts as Tumor Suppressor in Breast Cancer.Cancers (Basel). 2019 Jun 8;11(6):797. doi: 10.3390/cancers11060797. Cancers (Basel). 2019. PMID: 31181813 Free PMC article.
-
DNA Methylation Analysis of Ribosomal DNA in Adults With Down Syndrome.Front Genet. 2022 Apr 27;13:792165. doi: 10.3389/fgene.2022.792165. eCollection 2022. Front Genet. 2022. PMID: 35571061 Free PMC article.
-
Selective demethylation of two CpG sites causes postnatal activation of the Dao gene and consequent removal of D-serine within the mouse cerebellum.Clin Epigenetics. 2019 Oct 28;11(1):149. doi: 10.1186/s13148-019-0732-z. Clin Epigenetics. 2019. PMID: 31661019 Free PMC article.
-
Epigenetic remodelling of Fxyd1 promoters in developing heart and brain tissues.Sci Rep. 2022 Apr 19;12(1):6471. doi: 10.1038/s41598-022-10365-y. Sci Rep. 2022. PMID: 35440736 Free PMC article.
References
-
- Beygo J, Ammerpohl O, Gritzan D, Heitmann M, Rademacher K, Richter J, Caliebe A, Siebert R, Horsthemke B, Buiting K. Deep Bisulfite Sequencing of Aberrantly Methylated Loci in a Patient with Multiple Methylation Defects. PLoS One. 2013;8(10):e76953. doi: 10.1371/journal.pone.0076953. - DOI - PMC - PubMed
-
- Landan G, Cohen N, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein D, Goldfinger N, Zundelevich A, et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 2012;44(11):1207–1214. doi: 10.1038/ng.2442. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
