

Negative Allosteric Modulation of mGluR5 Partially Corrects Pathophysiology in a Mouse Model of Rett Syndrome
- PMID: 27881780
- PMCID: PMC5125247
- DOI: 10.1523/JNEUROSCI.0672-16.2016
Negative Allosteric Modulation of mGluR5 Partially Corrects Pathophysiology in a Mouse Model of Rett Syndrome
Abstract
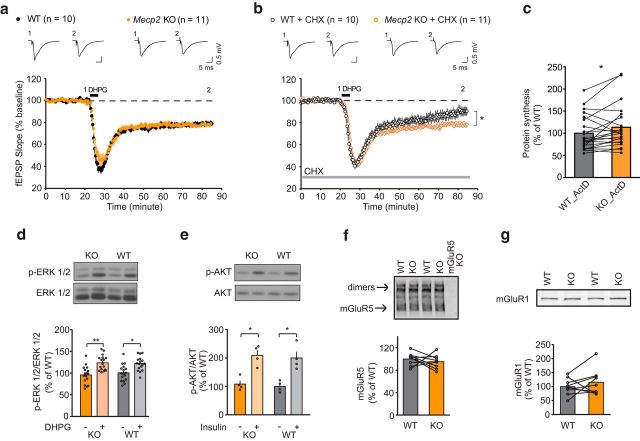
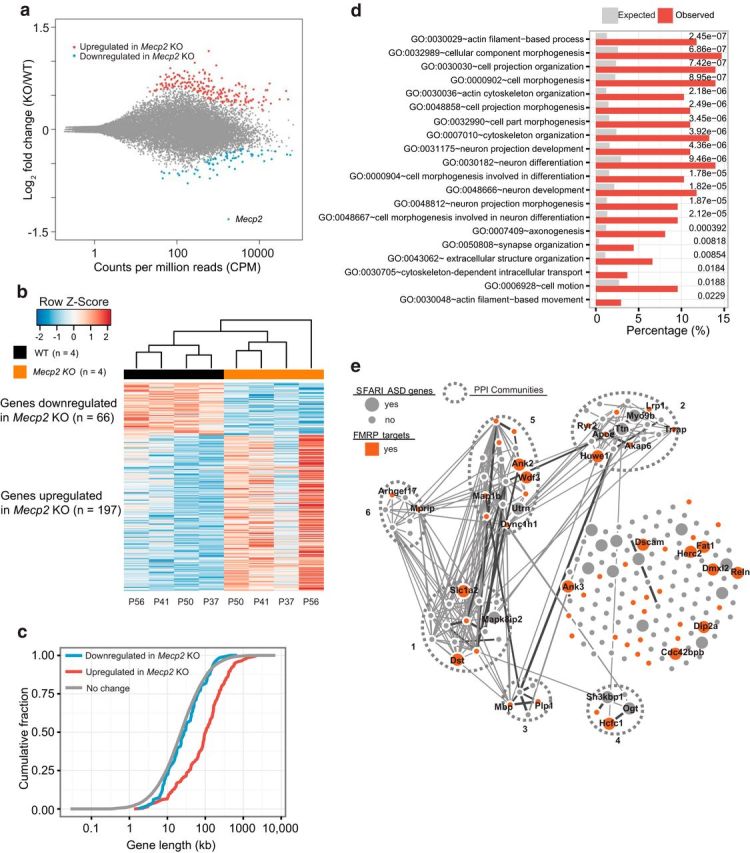
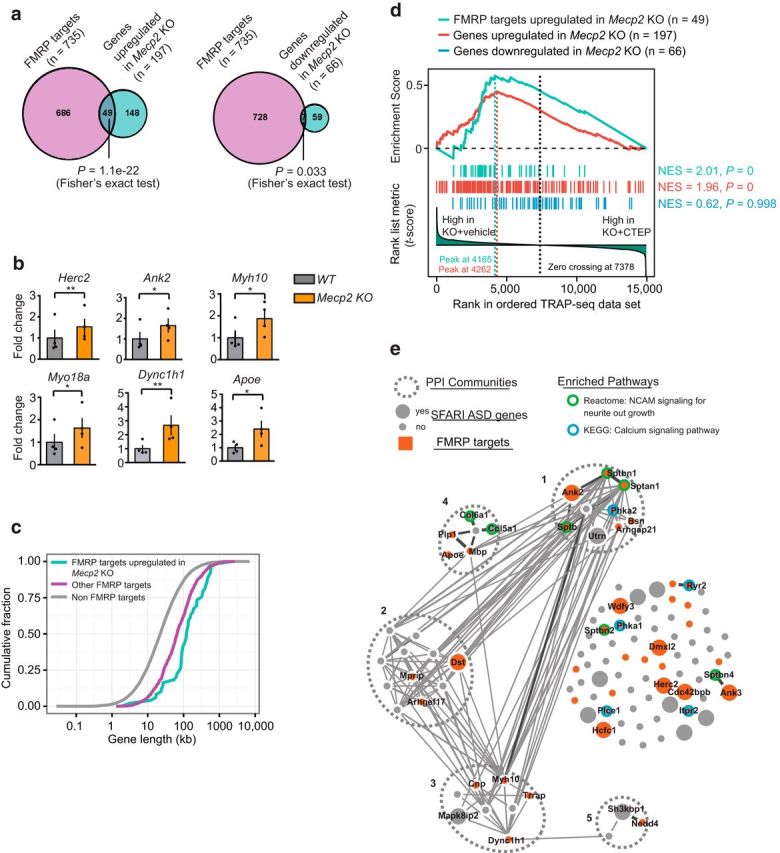
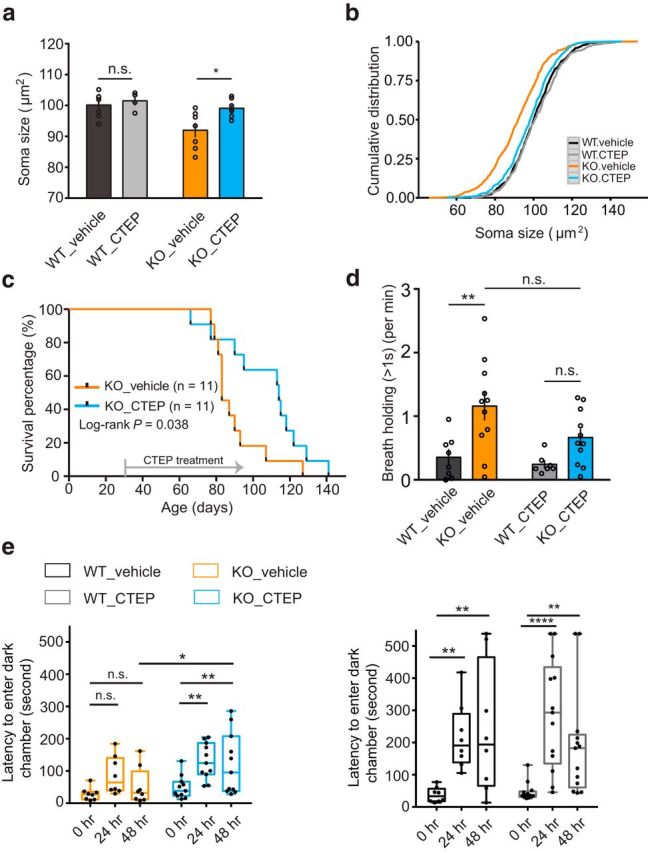
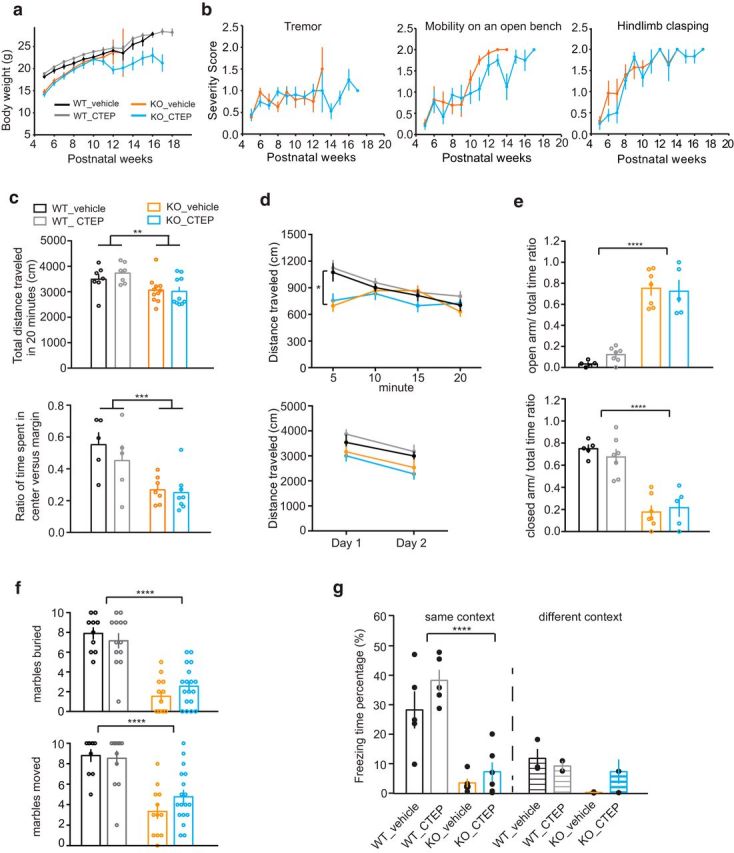
Rett syndrome (RTT) is caused by mutations in the gene encoding methyl-CpG binding protein 2 (MECP2), an epigenetic regulator of mRNA transcription. Here, we report a test of the hypothesis of shared pathophysiology of RTT and fragile X, another monogenic cause of autism and intellectual disability. In fragile X, the loss of the mRNA translational repressor FMRP leads to exaggerated protein synthesis downstream of metabotropic glutamate receptor 5 (mGluR5). We found that mGluR5- and protein-synthesis-dependent synaptic plasticity were similarly altered in area CA1 of Mecp2 KO mice. CA1 pyramidal cell-type-specific, genome-wide profiling of ribosome-bound mRNAs was performed in wild-type and Mecp2 KO hippocampal CA1 neurons to reveal the MeCP2-regulated "translatome." We found significant overlap between ribosome-bound transcripts overexpressed in the Mecp2 KO and FMRP mRNA targets. These tended to encode long genes that were functionally related to either cytoskeleton organization or the development of neuronal connectivity. In the Fmr1 KO mouse, chronic treatment with mGluR5-negative allosteric modulators (NAMs) has been shown to ameliorate many mutant phenotypes by correcting excessive protein synthesis. In Mecp2 KO mice, we found that mGluR5 NAM treatment significantly reduced the level of overexpressed ribosome-associated transcripts, particularly those that were also FMRP targets. Some Rett phenotypes were also ameliorated by treatment, most notably hippocampal cell size and lifespan. Together, these results suggest a potential mechanistic link between MeCP2-mediated transcription regulation and mGluR5/FMRP-mediated protein translation regulation through coregulation of a subset of genes relevant to synaptic functions.
Significance statement: Altered regulation of synaptic protein synthesis has been hypothesized to contribute to the pathophysiology that underlies multiple forms of intellectual disability and autism spectrum disorder. Here, we show in a mouse model of Rett syndrome (Mecp2 KO) that metabotropic glutamate receptor 5 (mGluR5)- and protein-synthesis-dependent synaptic plasticity are abnormal in the hippocampus. We found that a subset of ribosome-bound mRNAs was aberrantly upregulated in hippocampal CA1 neurons of Mecp2 KO mice, that these significantly overlapped with FMRP direct targets and/or SFARI human autism genes, and that chronic treatment of Mecp2 KO mice with an mGluR5-negative allosteric modulator tunes down upregulated ribosome-bound mRNAs and partially improves mutant mice phenotypes.
Keywords: Rett syndrome; autism; fragile X; intellectual disability; metabotropic glutamate receptor; synaptic protein synthesis.
Copyright © 2016 the authors 0270-6474/16/3611946-13$15.00/0.
Figures
Similar articles
-
EEA1 restores homeostatic synaptic plasticity in hippocampal neurons from Rett syndrome mice.J Physiol. 2017 Aug 15;595(16):5699-5712. doi: 10.1113/JP274450. Epub 2017 Jul 12. J Physiol. 2017. PMID: 28621434 Free PMC article.
-
mGlu5 positive allosteric modulation normalizes synaptic plasticity defects and motor phenotypes in a mouse model of Rett syndrome.Hum Mol Genet. 2016 May 15;25(10):1990-2004. doi: 10.1093/hmg/ddw074. Epub 2016 Mar 2. Hum Mol Genet. 2016. PMID: 26936821 Free PMC article.
-
Effects of the presence and absence of amino acids on translation, signaling, and long-term depression in hippocampal slices from Fmr1 knockout mice.J Neurochem. 2019 Dec;151(6):764-776. doi: 10.1111/jnc.14874. Epub 2019 Nov 12. J Neurochem. 2019. PMID: 31539452 Free PMC article.
-
Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development.Psychopharmacology (Berl). 2014 Mar;231(6):1217-26. doi: 10.1007/s00213-013-3330-3. Psychopharmacology (Berl). 2014. PMID: 24232444 Review.
-
Rett Syndrome and Fragile X Syndrome: Different Etiology With Common Molecular Dysfunctions.Front Cell Neurosci. 2021 Nov 19;15:764761. doi: 10.3389/fncel.2021.764761. eCollection 2021. Front Cell Neurosci. 2021. PMID: 34867203 Free PMC article. Review.
Cited by
-
Sex-Specific Impact of Fkbp5 on Hippocampal Response to Acute Alcohol Injection: Involvement in Alterations of Metabolism-Related Pathways.Cells. 2023 Dec 31;13(1):89. doi: 10.3390/cells13010089. Cells. 2023. PMID: 38201293 Free PMC article.
-
SHANK2 Mutations Result in Dysregulation of the ERK1/2 Pathway in Human Induced Pluripotent Stem Cells-Derived Neurons and Shank2(-/-) Mice.Front Mol Neurosci. 2021 Nov 26;14:773571. doi: 10.3389/fnmol.2021.773571. eCollection 2021. Front Mol Neurosci. 2021. PMID: 34899182 Free PMC article.
-
Modernising autism spectrum disorder model engineering and treatment via CRISPR-Cas9: A gene reprogramming approach.World J Clin Cases. 2023 May 16;11(14):3114-3127. doi: 10.12998/wjcc.v11.i14.3114. World J Clin Cases. 2023. PMID: 37274051 Free PMC article. Review.
-
Role of mGlu5 in Persistent Forms of Hippocampal Synaptic Plasticity and the Encoding of Spatial Experience.Cells. 2022 Oct 24;11(21):3352. doi: 10.3390/cells11213352. Cells. 2022. PMID: 36359749 Free PMC article. Review.
-
Translatome Regulation in Neuronal Injury and Axon Regrowth.eNeuro. 2018 May 10;5(2):ENEURO.0276-17.2018. doi: 10.1523/ENEURO.0276-17.2018. eCollection 2018 Mar-Apr. eNeuro. 2018. PMID: 29756027 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
