

Centrifuge: rapid and sensitive classification of metagenomic sequences
- PMID: 27852649
- PMCID: PMC5131823
- DOI: 10.1101/gr.210641.116
Centrifuge: rapid and sensitive classification of metagenomic sequences
Abstract
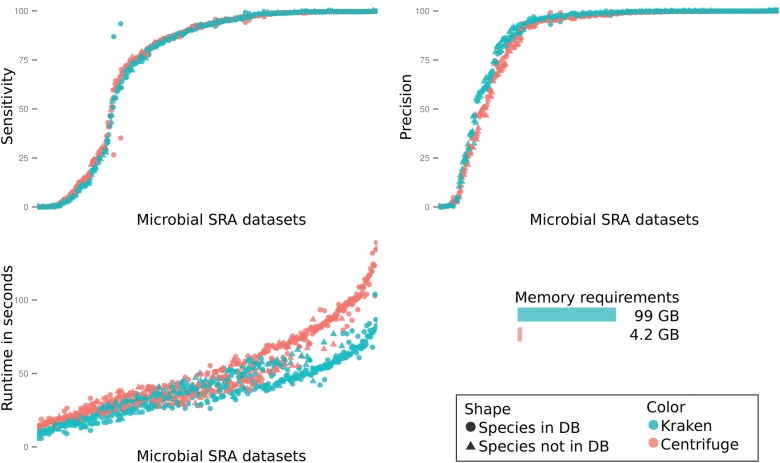
Centrifuge is a novel microbial classification engine that enables rapid, accurate, and sensitive labeling of reads and quantification of species on desktop computers. The system uses an indexing scheme based on the Burrows-Wheeler transform (BWT) and the Ferragina-Manzini (FM) index, optimized specifically for the metagenomic classification problem. Centrifuge requires a relatively small index (4.2 GB for 4078 bacterial and 200 archaeal genomes) and classifies sequences at very high speed, allowing it to process the millions of reads from a typical high-throughput DNA sequencing run within a few minutes. Together, these advances enable timely and accurate analysis of large metagenomics data sets on conventional desktop computers. Because of its space-optimized indexing schemes, Centrifuge also makes it possible to index the entire NCBI nonredundant nucleotide sequence database (a total of 109 billion bases) with an index size of 69 GB, in contrast to k-mer-based indexing schemes, which require far more extensive space.
© 2016 Kim et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
CLAST: CUDA implemented large-scale alignment search tool.BMC Bioinformatics. 2014 Dec 11;15(1):406. doi: 10.1186/s12859-014-0406-y. BMC Bioinformatics. 2014. PMID: 25495907 Free PMC article.
-
Fast and sensitive taxonomic classification for metagenomics with Kaiju.Nat Commun. 2016 Apr 13;7:11257. doi: 10.1038/ncomms11257. Nat Commun. 2016. PMID: 27071849 Free PMC article.
-
A framework for space-efficient read clustering in metagenomic samples.BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):59. doi: 10.1186/s12859-017-1466-6. BMC Bioinformatics. 2017. PMID: 28361710 Free PMC article.
-
Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques.Antonie Van Leeuwenhoek. 2017 Oct;110(10):1357-1371. doi: 10.1007/s10482-017-0928-1. Epub 2017 Aug 22. Antonie Van Leeuwenhoek. 2017. PMID: 28831610 Review.
-
From genomics to metagenomics.Curr Opin Biotechnol. 2012 Feb;23(1):72-6. doi: 10.1016/j.copbio.2011.12.017. Epub 2012 Jan 5. Curr Opin Biotechnol. 2012. PMID: 22227326 Review.
Cited by
-
Metagenomics: A viable tool for reconstructing herbivore diet.Mol Ecol Resour. 2021 Oct;21(7):2249-2263. doi: 10.1111/1755-0998.13425. Epub 2021 May 25. Mol Ecol Resour. 2021. PMID: 33971086 Free PMC article.
-
The Impact of Migration on the Gut Metagenome of South Asian Canadians.Gut Microbes. 2021 Jan-Dec;13(1):1-29. doi: 10.1080/19490976.2021.1902705. Gut Microbes. 2021. PMID: 33794735 Free PMC article.
-
Liver transcriptome resources of four commercially exploited teleost species.Sci Data. 2020 Jul 7;7(1):214. doi: 10.1038/s41597-020-0565-9. Sci Data. 2020. PMID: 32636445 Free PMC article.
-
Microbial functional pathways based on metatranscriptomic profiling enable effective saliva-based health assessments for precision wellness.Comput Struct Biotechnol J. 2024 Jan 29;23:834-842. doi: 10.1016/j.csbj.2024.01.018. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38328005 Free PMC article.
-
DNA Thermo-Protection Facilitates Whole-Genome Sequencing of Mycobacteria Direct from Clinical Samples.J Clin Microbiol. 2020 Sep 22;58(10):e00670-20. doi: 10.1128/JCM.00670-20. Print 2020 Sep 22. J Clin Microbiol. 2020. PMID: 32719032 Free PMC article.
References
-
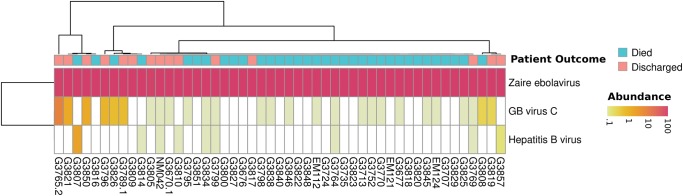
- Baize S, Pannetier D, Oestereich L, Rieger T, Koivogui L, Magassouba N, Soropogui B, Sow MS, Keita S, De Clerck H, et al. 2014. Emergence of Zaire Ebola virus disease in Guinea. N Engl J Med 371: 1418–1425. - PubMed
-
- Burrows M, Wheeler DJ. 1994. A block-sorting lossless data compression algorithm. Technical Report 124 Digital Equipment Corporation, Palo Alto, CA.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous