

WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) Inhibits GLUT1-mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site
- PMID: 27836974
- PMCID: PMC5207184
- DOI: 10.1074/jbc.M116.759175
WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) Inhibits GLUT1-mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site
Abstract
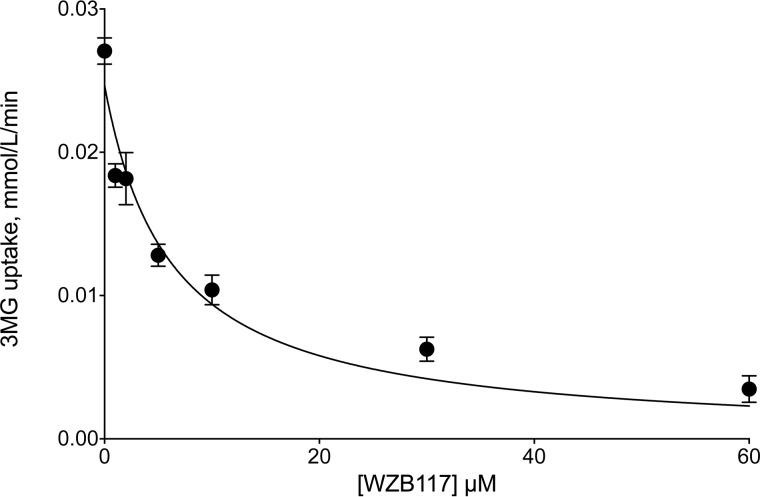
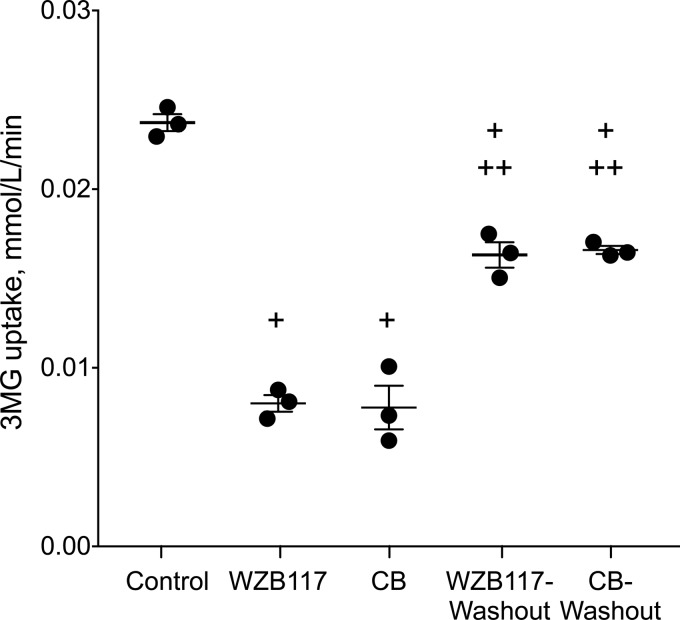
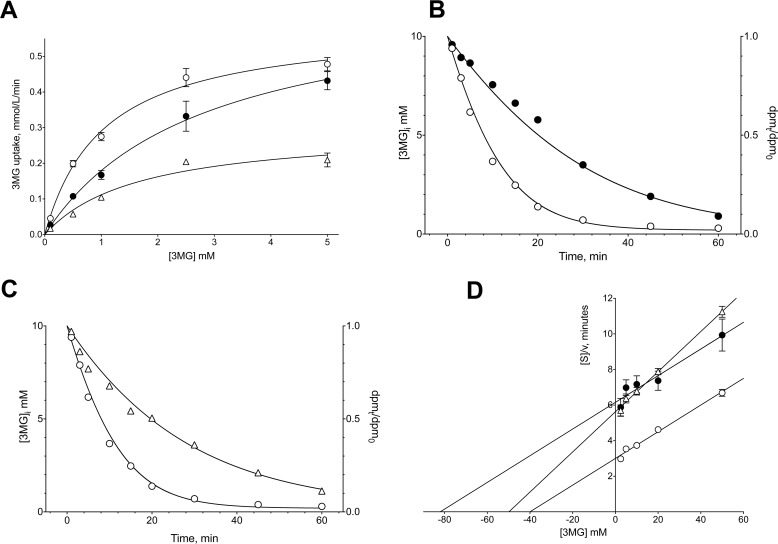
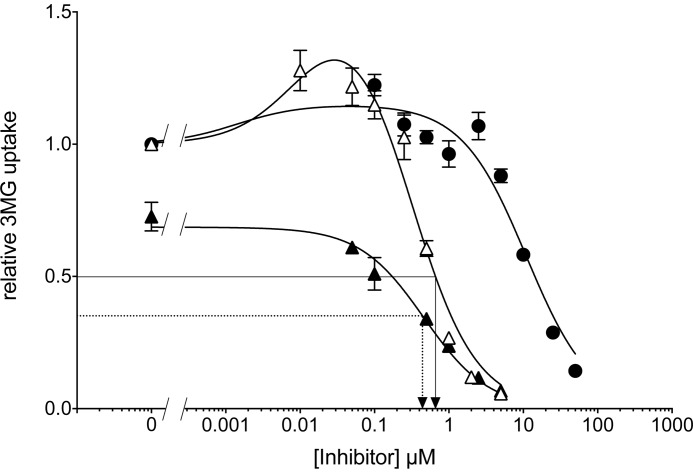
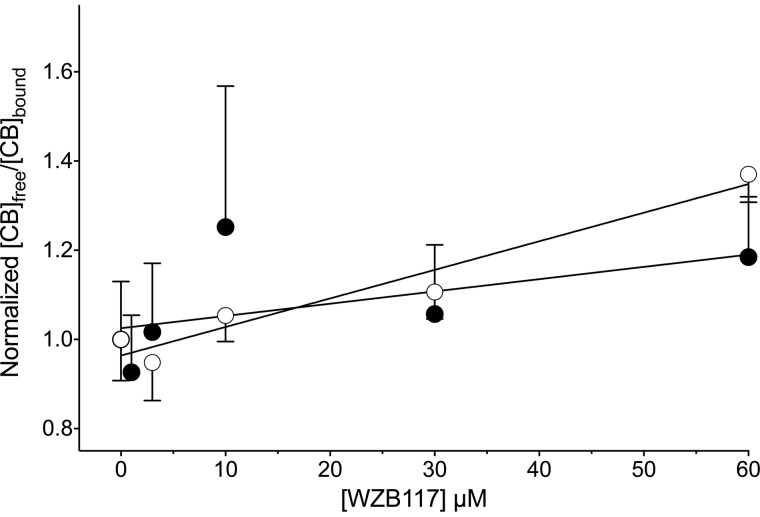
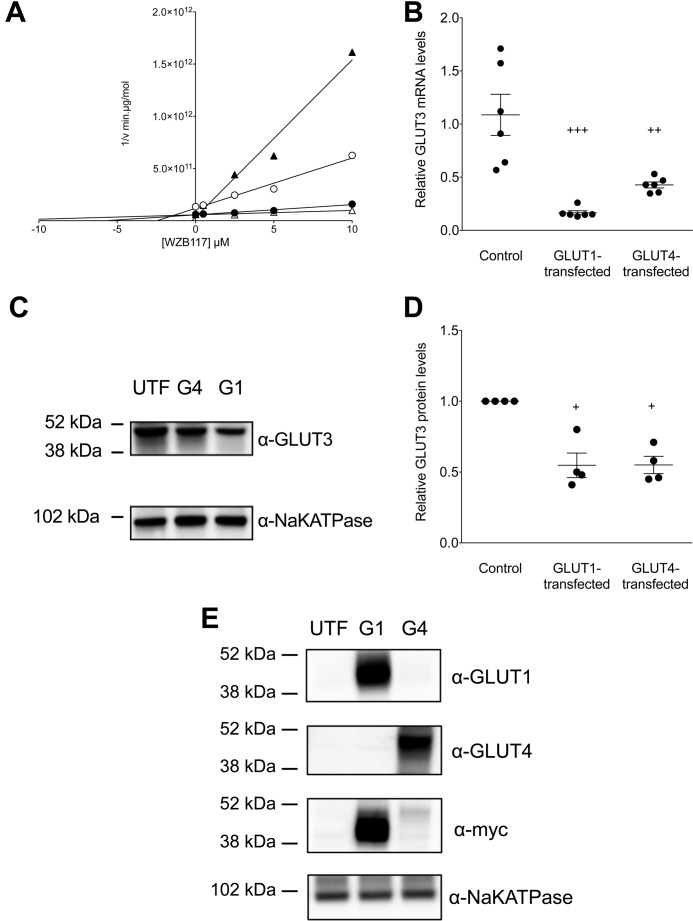
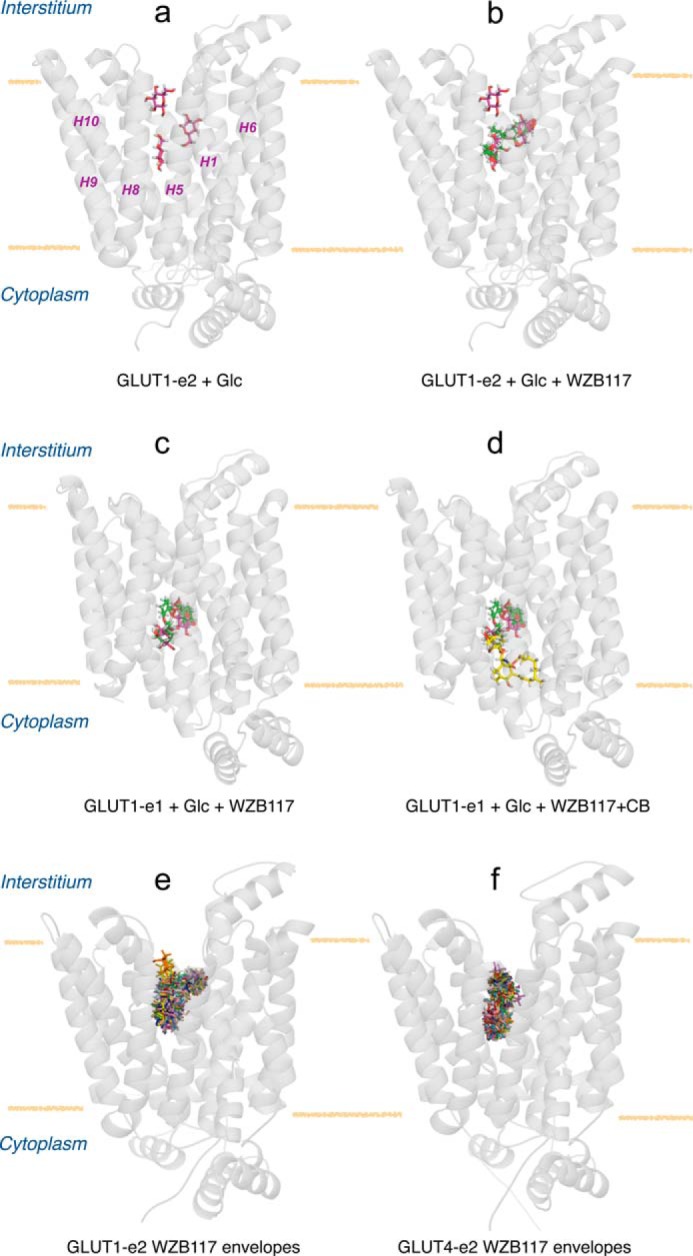
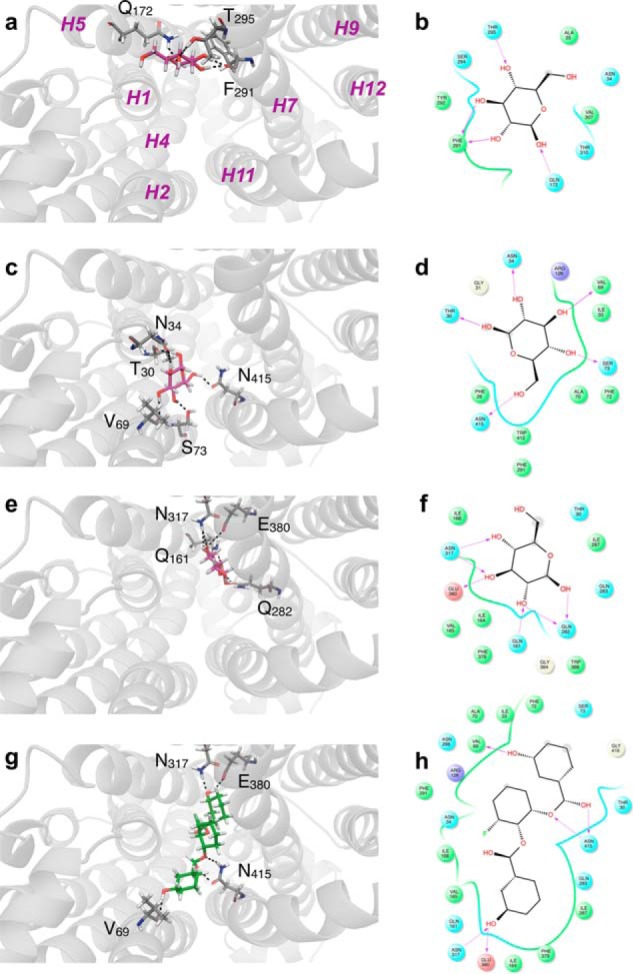
WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate) inhibits passive sugar transport in human erythrocytes and cancer cell lines and, by limiting glycolysis, inhibits tumor growth in mice. This study explores how WZB117 inhibits the erythrocyte sugar transporter glucose transport protein 1 (GLUT1) and examines the transporter isoform specificity of inhibition. WZB117 reversibly and competitively inhibits erythrocyte 3-O-methylglucose (3MG) uptake with Ki(app) = 6 μm but is a noncompetitive inhibitor of sugar exit. Cytochalasin B (CB) is a reversible, noncompetitive inhibitor of 3MG uptake with Ki(app) = 0.3 μm but is a competitive inhibitor of sugar exit indicating that WZB117 and CB bind at exofacial and endofacial sugar binding sites, respectively. WZB117 inhibition of GLUTs expressed in HEK293 cells follows the order of potency: insulin-regulated GLUT4 ≫ GLUT1 ≈ neuronal GLUT3. This may explain WZB117-induced murine lipodystrophy. Molecular docking suggests the following. 1) The WZB117 binding envelopes of exofacial GLUT1 and GLUT4 conformers differ significantly. 2) GLUT1 and GLUT4 exofacial conformers present multiple, adjacent glucose binding sites that overlap with WZB117 binding envelopes. 3) The GLUT1 exofacial conformer lacks a CB binding site. 4) The inward GLUT1 conformer presents overlapping endofacial WZB117, d-glucose, and CB binding envelopes. Interrogating the GLUT1 mechanism using WZB117 reveals that subsaturating WZB117 and CB stimulate erythrocyte 3MG uptake. Extracellular WZB117 does not affect CB binding to GLUT1, but intracellular WZB117 inhibits CB binding. These findings are incompatible with the alternating conformer carrier for glucose transport but are consistent with either a multisubunit, allosteric transporter, or a transporter in which each subunit presents multiple, interacting ligand binding sites.
Keywords: competitive inhibition; facilitated diffusion; glucose transport; ligand binding; ligand-binding protein; membrane transport; membrane transport protein; molecular docking; protein structure.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Rapid substrate translocation by the multisubunit, erythroid glucose transporter requires subunit associations but not cooperative ligand binding.Biochemistry. 1995 Aug 1;34(30):9762-73. doi: 10.1021/bi00030a014. Biochemistry. 1995. PMID: 7626647
-
Caffeine inhibits glucose transport by binding at the GLUT1 nucleotide-binding site.Am J Physiol Cell Physiol. 2015 May 15;308(10):C827-34. doi: 10.1152/ajpcell.00001.2015. Epub 2015 Feb 25. Am J Physiol Cell Physiol. 2015. PMID: 25715702 Free PMC article.
-
Determinants of ligand binding affinity and cooperativity at the GLUT1 endofacial site.Biochemistry. 2011 Apr 19;50(15):3137-48. doi: 10.1021/bi1020327. Epub 2011 Mar 25. Biochemistry. 2011. PMID: 21384913 Free PMC article.
-
Will the original glucose transporter isoform please stand up!Am J Physiol Endocrinol Metab. 2009 Oct;297(4):E836-48. doi: 10.1152/ajpendo.00496.2009. Epub 2009 Aug 18. Am J Physiol Endocrinol Metab. 2009. PMID: 19690067 Free PMC article. Review.
-
Erythroid glucose transport in health and disease.Pflugers Arch. 2020 Sep;472(9):1371-1383. doi: 10.1007/s00424-020-02406-0. Epub 2020 May 30. Pflugers Arch. 2020. PMID: 32474749 Review.
Cited by
-
A small-molecule pan-class I glucose transporter inhibitor reduces cancer cell proliferation in vitro and tumor growth in vivo by targeting glucose-based metabolism.Cancer Metab. 2021 Mar 26;9(1):14. doi: 10.1186/s40170-021-00248-7. Cancer Metab. 2021. PMID: 33771231 Free PMC article.
-
The Intratumoral Heterogeneity of Cancer Metabolism.Adv Exp Med Biol. 2018;1063:131-145. doi: 10.1007/978-3-319-77736-8_10. Adv Exp Med Biol. 2018. PMID: 29946781 Review.
-
Development of Glucose Transporter (GLUT) Inhibitors.European J Org Chem. 2020 May 3;2020(16):2321-2329. doi: 10.1002/ejoc.201901353. Epub 2019 Nov 28. European J Org Chem. 2020. PMID: 32421048 Free PMC article. Review.
-
The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition.Nat Commun. 2017 May 26;8:15503. doi: 10.1038/ncomms15503. Nat Commun. 2017. PMID: 28548087 Free PMC article.
-
Reconciling contradictory findings: Glucose transporter 1 (GLUT1) functions as an oligomer of allosteric, alternating access transporters.J Biol Chem. 2017 Dec 22;292(51):21035-21046. doi: 10.1074/jbc.M117.815589. Epub 2017 Oct 24. J Biol Chem. 2017. PMID: 29066623 Free PMC article.
References
-
- Palorini R., Cammarata F. P., Cammarata F., Balestrieri C., Monestiroli A., Vasso M., Gelfi C., Alberghina L., and Chiaradonna F. (2013) Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death Dis. 4, e732. - PMC - PubMed
-
- Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 - PubMed
-
- De Giorgis V., and Veggiotti P. (2013) GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22, 803–811 - PubMed
-
- Pelicano H., Martin D. S., Xu R. H., and Huang P. (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
