

Arabidopsis thaliana population analysis reveals high plasticity of the genomic region spanning MSH2, AT3G18530 and AT3G18535 genes and provides evidence for NAHR-driven recurrent CNV events occurring in this location
- PMID: 27825302
- PMCID: PMC5101643
- DOI: 10.1186/s12864-016-3221-1
Arabidopsis thaliana population analysis reveals high plasticity of the genomic region spanning MSH2, AT3G18530 and AT3G18535 genes and provides evidence for NAHR-driven recurrent CNV events occurring in this location
Abstract
Background: Intraspecies copy number variations (CNVs), defined as unbalanced structural variations of specific genomic loci, ≥1 kb in size, are present in the genomes of animals and plants. A growing number of examples indicate that CNVs may have functional significance and contribute to phenotypic diversity. In the model plant Arabidopsis thaliana at least several hundred protein-coding genes might display CNV; however, locus-specific genotyping studies in this plant have not been conducted.
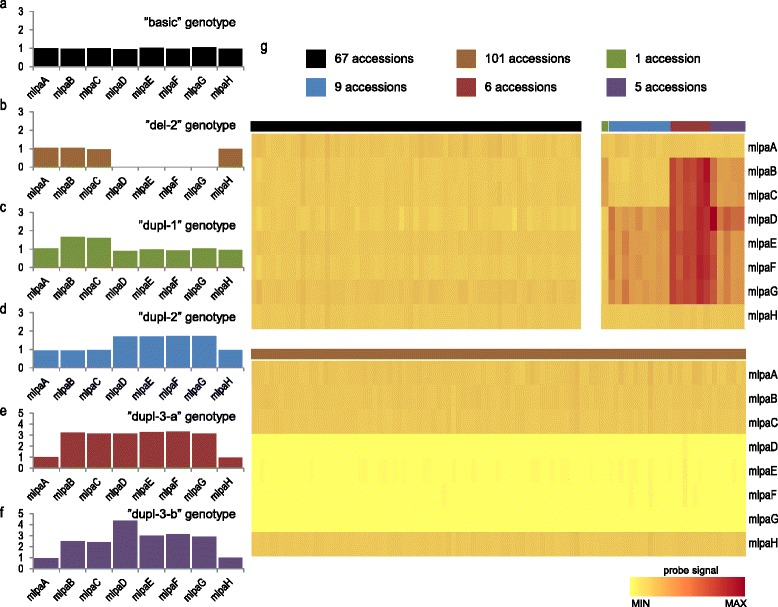
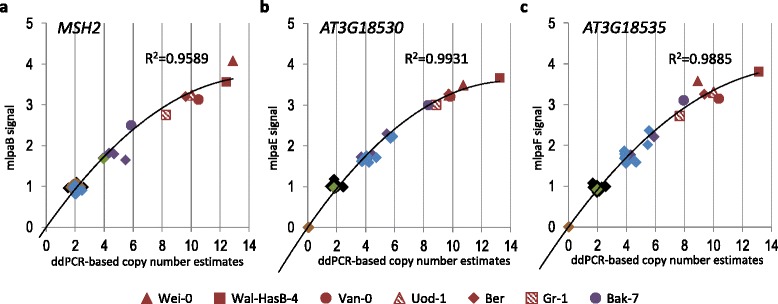
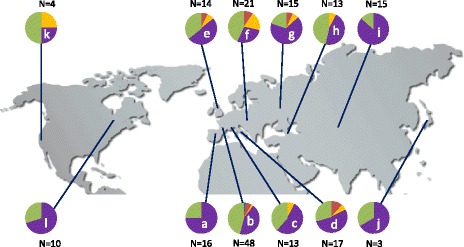
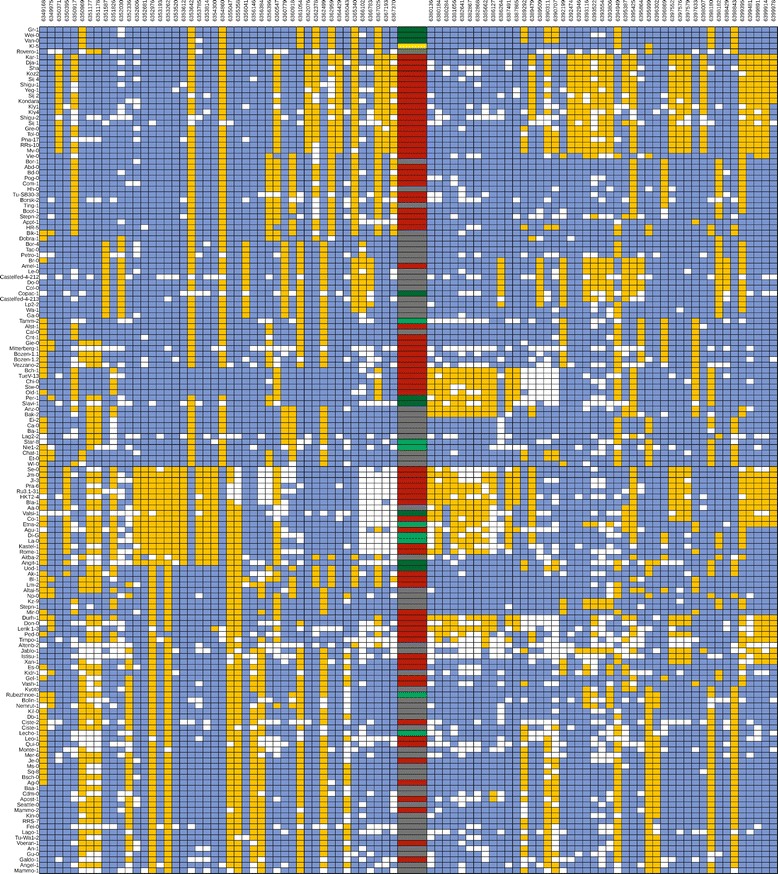
Results: We analyzed the natural CNVs in the region overlapping MSH2 gene that encodes the DNA mismatch repair protein, and AT3G18530 and AT3G18535 genes that encode poorly characterized proteins. By applying multiplex ligation-dependent probe amplification and droplet digital PCR we genotyped those genes in 189 A. thaliana accessions. We found that AT3G18530 and AT3G18535 were duplicated (2-14 times) in 20 and deleted in 101 accessions. MSH2 was duplicated in 12 accessions (up to 12-14 copies) but never deleted. In all but one case, the MSH2 duplications were associated with those of AT3G18530 and AT3G18535. Considering the structure of the CNVs, we distinguished 5 genotypes for this region, determined their frequency and geographical distribution. We defined the CNV breakpoints in 35 accessions with AT3G18530 and AT3G18535 deletions and tandem duplications and showed that they were reciprocal events, resulting from non-allelic homologous recombination between 99 %-identical sequences flanking these genes. The widespread geographical distribution of the deletions supported by the SNP and linkage disequilibrium analyses of the genomic sequence confirmed the recurrent nature of this CNV.
Conclusions: We characterized in detail for the first time the complex multiallelic CNV in Arabidopsis genome. The region encoding MSH2, AT3G18530 and AT3G18535 genes shows enormous variation of copy numbers among natural ecotypes, being a remarkable example of high Arabidopsis genome plasticity. We provided the molecular insight into the mechanism underlying the recurrent nature of AT3G18530-AT3G18535 duplications/deletions. We also performed the first direct comparison of the two leading experimental methods, suitable for assessing the DNA copy number status. Our comprehensive case study provides foundation information for further analyses of CNV evolution in Arabidopsis and other plants, and their possible use in plant breeding.
Keywords: Arabidopsis thaliana; Copy number variation (CNV); Droplet digital PCR; Genotyping; Multiallelic CNV; Multiplex ligation-dependent probe amplification (MLPA); Non-allelic homologous recombination (NAHR); Recurrent deletion.
Figures




Similar articles
-
AthCNV: A Map of DNA Copy Number Variations in the Arabidopsis Genome.Plant Cell. 2020 Jun;32(6):1797-1819. doi: 10.1105/tpc.19.00640. Epub 2020 Apr 7. Plant Cell. 2020. PMID: 32265262 Free PMC article.
-
Correlation between frequency of non-allelic homologous recombination and homology properties: evidence from homology-mediated CNV mutations in the human genome.Hum Mol Genet. 2015 Mar 1;24(5):1225-33. doi: 10.1093/hmg/ddu533. Epub 2014 Oct 16. Hum Mol Genet. 2015. PMID: 25324539
-
MLPA-Based Analysis of Copy Number Variation in Plant Populations.Front Plant Sci. 2017 Feb 21;8:222. doi: 10.3389/fpls.2017.00222. eCollection 2017. Front Plant Sci. 2017. PMID: 28270823 Free PMC article.
-
Origins and breakpoint analyses of copy number variations: up close and personal.Cytogenet Genome Res. 2011;135(3-4):271-6. doi: 10.1159/000330267. Epub 2011 Aug 12. Cytogenet Genome Res. 2011. PMID: 21846967 Review.
-
Copy number polymorphism in plant genomes.Theor Appl Genet. 2014 Jan;127(1):1-18. doi: 10.1007/s00122-013-2177-7. Epub 2013 Aug 29. Theor Appl Genet. 2014. PMID: 23989647 Free PMC article. Review.
Cited by
-
Copy Number Variation among Resistance Genes Analogues in Brassica napus.Genes (Basel). 2022 Nov 4;13(11):2037. doi: 10.3390/genes13112037. Genes (Basel). 2022. PMID: 36360273 Free PMC article.
-
Molecular Characterization and Functional Analysis of the Hb-hsp90-1 Gene in Relation to Temperature Changes in Heterorhabditis bacteriophora.Front Physiol. 2021 Feb 23;12:615653. doi: 10.3389/fphys.2021.615653. eCollection 2021. Front Physiol. 2021. PMID: 33732162 Free PMC article.
-
Digital PCR: What Relevance to Plant Studies?Biology (Basel). 2020 Nov 30;9(12):433. doi: 10.3390/biology9120433. Biology (Basel). 2020. PMID: 33266157 Free PMC article. Review.
-
AthCNV: A Map of DNA Copy Number Variations in the Arabidopsis Genome.Plant Cell. 2020 Jun;32(6):1797-1819. doi: 10.1105/tpc.19.00640. Epub 2020 Apr 7. Plant Cell. 2020. PMID: 32265262 Free PMC article.
-
Gynoecy instability in cucumber (Cucumis sativus L.) is due to unequal crossover at the copy number variation-dependent Femaleness (F) locus.Hortic Res. 2020 Mar 15;7:32. doi: 10.1038/s41438-020-0251-2. eCollection 2020. Hortic Res. 2020. PMID: 32194968 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
