

PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation
- PMID: 27779186
- PMCID: PMC5093342
- DOI: 10.1038/ncomms13280
PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation
Abstract
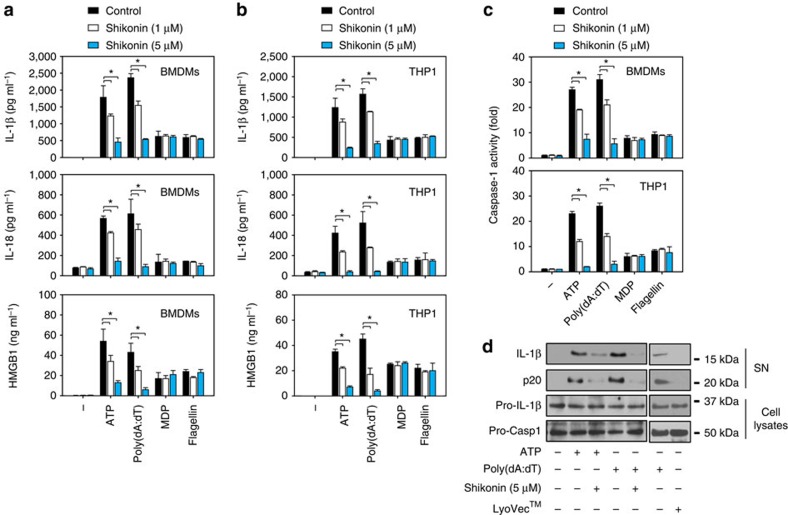
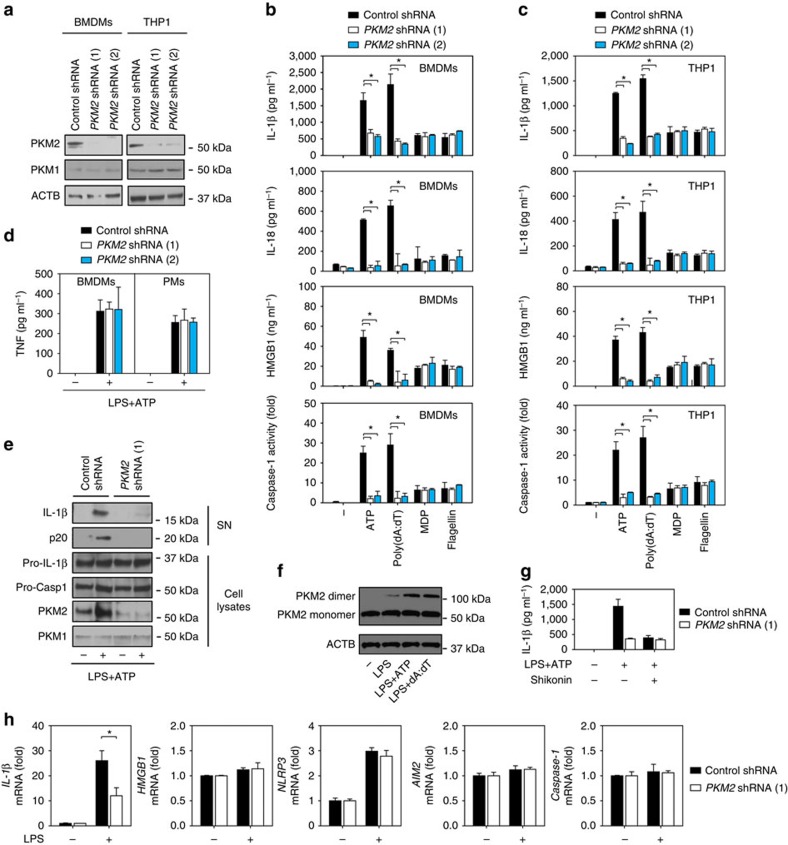
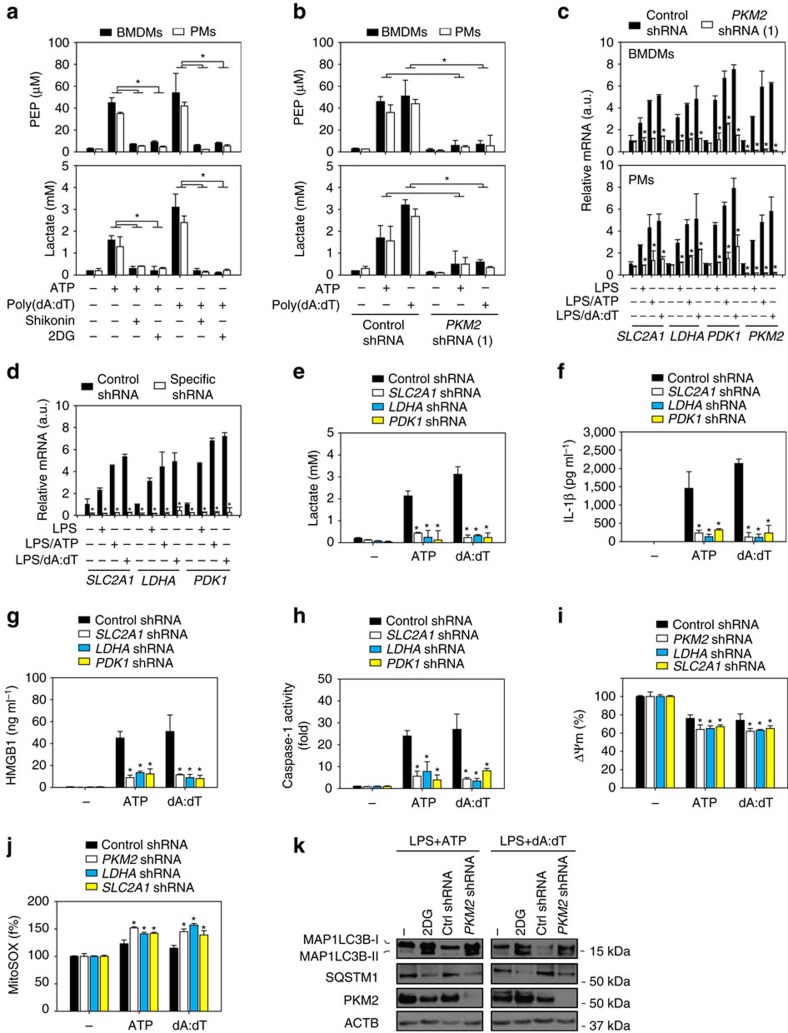
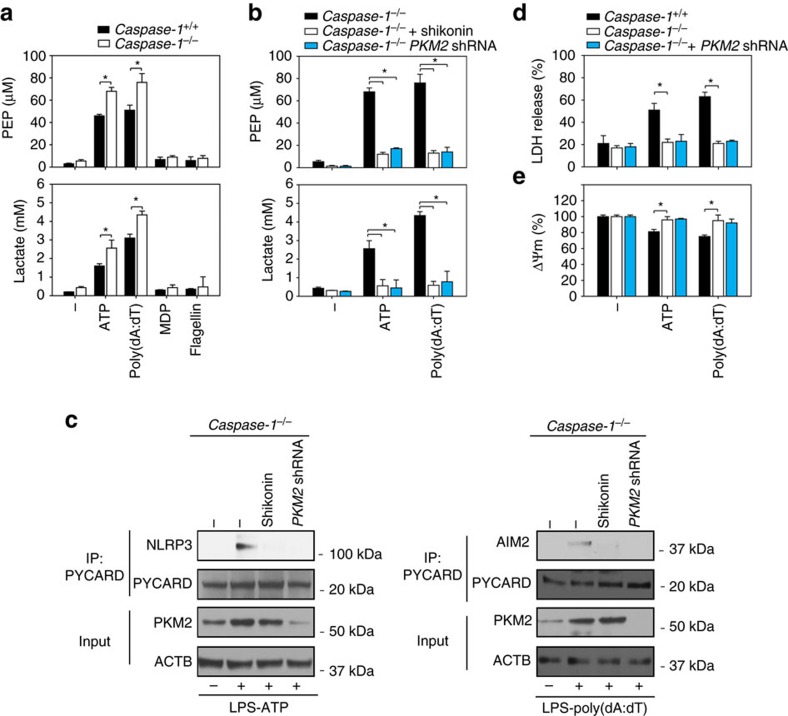
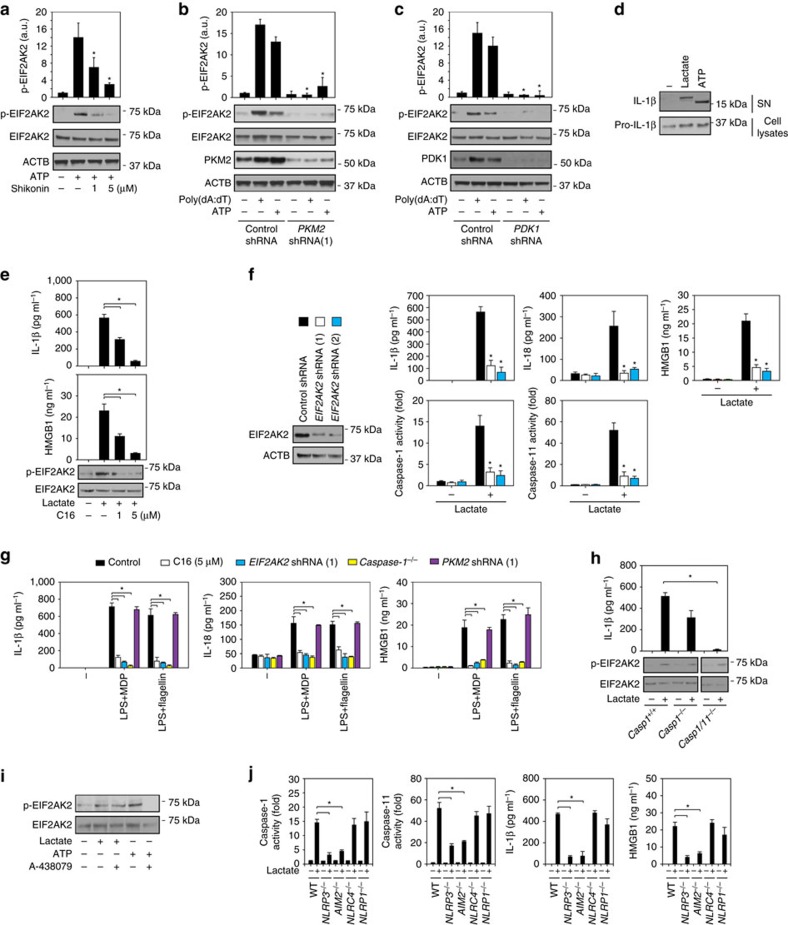
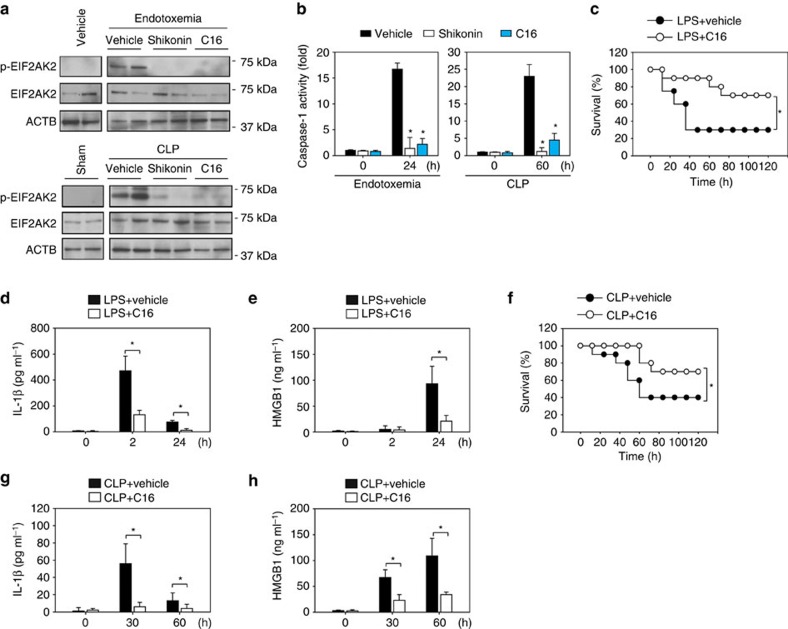
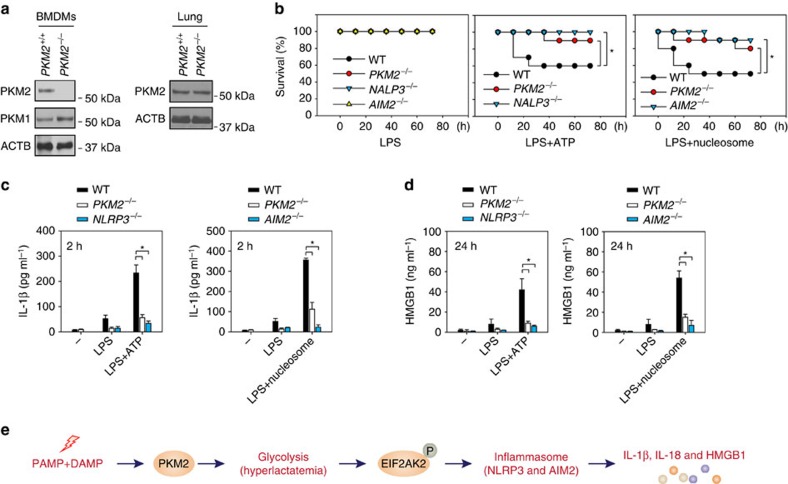
Sepsis, severe sepsis and septic shock are the main cause of mortality in non-cardiac intensive care units. Immunometabolism has been linked to sepsis; however, the precise mechanism by which metabolic reprogramming regulates the inflammatory response is unclear. Here we show that aerobic glycolysis contributes to sepsis by modulating inflammasome activation in macrophages. PKM2-mediated glycolysis promotes inflammasome activation by modulating EIF2AK2 phosphorylation in macrophages. Pharmacological and genetic inhibition of PKM2 or EIF2AK2 attenuates NLRP3 and AIM2 inflammasomes activation, and consequently suppresses the release of IL-1β, IL-18 and HMGB1 by macrophages. Pharmacological inhibition of the PKM2-EIF2AK2 pathway protects mice from lethal endotoxemia and polymicrobial sepsis. Moreover, conditional knockout of PKM2 in myeloid cells protects mice from septic death induced by NLRP3 and AIM2 inflammasome activation. These findings define an important role of PKM2 in immunometabolism and guide future development of therapeutic strategies to treat sepsis.
Conflict of interest statement
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
PKM2-dependent glycolysis promotes skeletal muscle cell pyroptosis by activating the NLRP3 inflammasome in dermatomyositis/polymyositis.Rheumatology (Oxford). 2021 May 14;60(5):2177-2189. doi: 10.1093/rheumatology/keaa473. Rheumatology (Oxford). 2021. PMID: 33165604
-
Novel role of PKR in inflammasome activation and HMGB1 release.Nature. 2012 Aug 30;488(7413):670-4. doi: 10.1038/nature11290. Nature. 2012. PMID: 22801494 Free PMC article.
-
PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis.Nat Commun. 2014 Jul 14;5:4436. doi: 10.1038/ncomms5436. Nat Commun. 2014. PMID: 25019241 Free PMC article.
-
Molecular mechanisms regulating NLRP3 inflammasome activation.Cell Mol Immunol. 2016 Mar;13(2):148-59. doi: 10.1038/cmi.2015.95. Epub 2015 Nov 9. Cell Mol Immunol. 2016. PMID: 26549800 Free PMC article. Review.
-
The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs.Int J Mol Sci. 2021 Jan 25;22(3):1171. doi: 10.3390/ijms22031171. Int J Mol Sci. 2021. PMID: 33503959 Free PMC article. Review.
Cited by
-
High iodine promotes autoimmune thyroid disease by activating hexokinase 3 and inducing polarization of macrophages towards M1.Front Immunol. 2022 Oct 17;13:1009932. doi: 10.3389/fimmu.2022.1009932. eCollection 2022. Front Immunol. 2022. PMID: 36325332 Free PMC article.
-
Dysregulation of Pyruvate Kinase M2 Promotes Inflammation in a Mouse Model of Obese Allergic Asthma.Am J Respir Cell Mol Biol. 2021 Jun;64(6):709-721. doi: 10.1165/rcmb.2020-0512OC. Am J Respir Cell Mol Biol. 2021. PMID: 33662229 Free PMC article.
-
The Role of the Pathogen Dose and PI3Kγ in Immunometabolic Reprogramming of Microglia for Innate Immune Memory.Int J Mol Sci. 2021 Mar 4;22(5):2578. doi: 10.3390/ijms22052578. Int J Mol Sci. 2021. PMID: 33806610 Free PMC article.
-
Autophagy-related biomarkers in preeclampsia: the underlying mechanism, correlation to the immune microenvironment and drug screening.BMC Pregnancy Childbirth. 2024 Jan 2;24(1):1. doi: 10.1186/s12884-023-06211-2. BMC Pregnancy Childbirth. 2024. PMID: 38166707 Free PMC article.
-
Glutamine metabolism modulates microglial NLRP3 inflammasome activity through mitophagy in Alzheimer's disease.J Neuroinflammation. 2024 Oct 15;21(1):261. doi: 10.1186/s12974-024-03254-w. J Neuroinflammation. 2024. PMID: 39407211 Free PMC article.
References
-
- Wang H. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
