

Defining synonymous codon compression schemes by genome recoding
- PMID: 27776354
- PMCID: PMC5321499
- DOI: 10.1038/nature20124
Defining synonymous codon compression schemes by genome recoding
Abstract
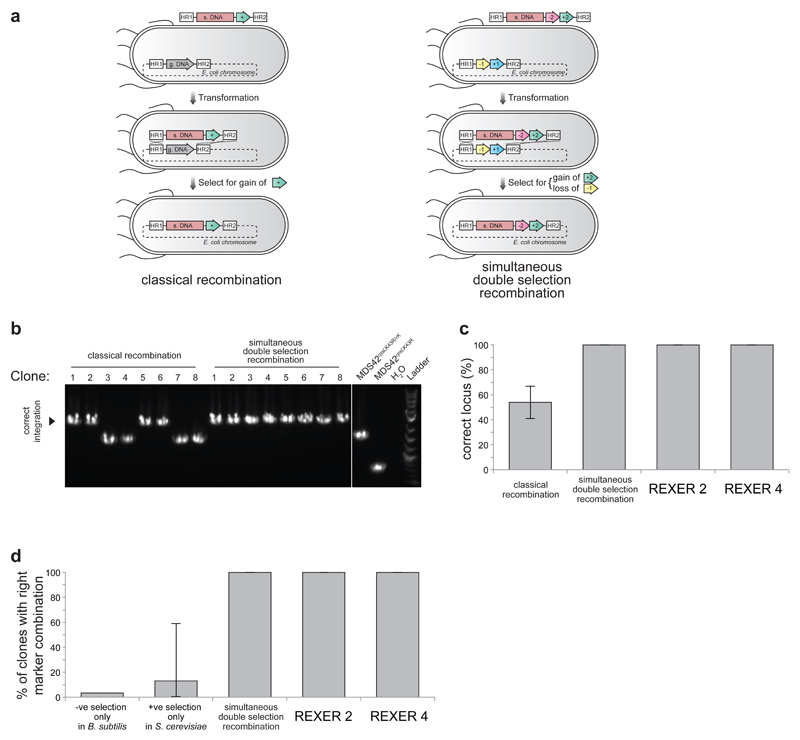
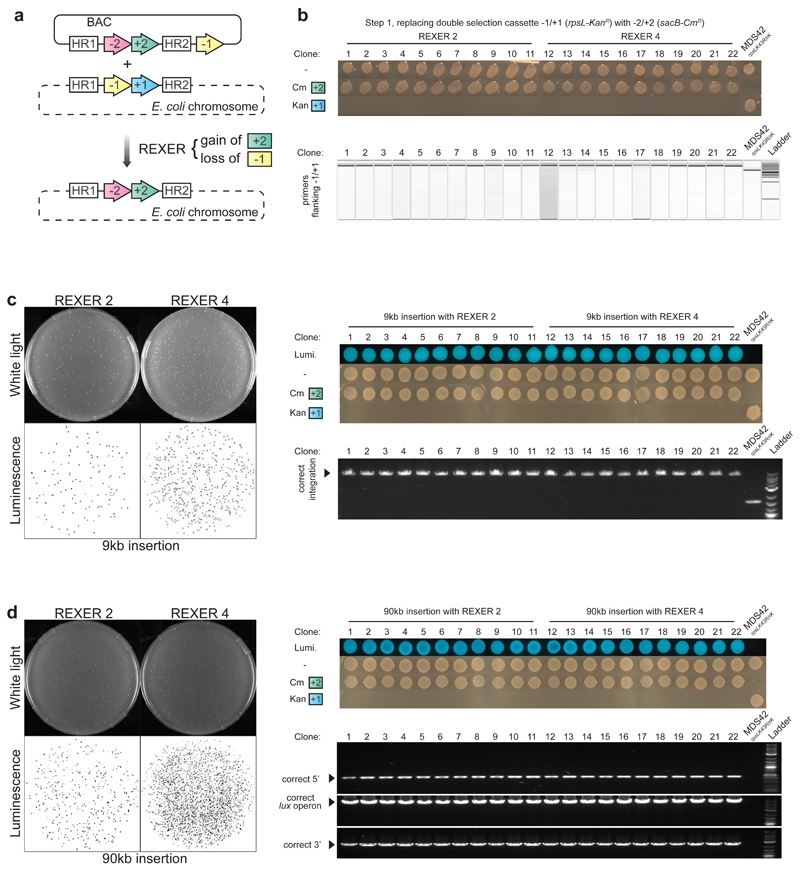
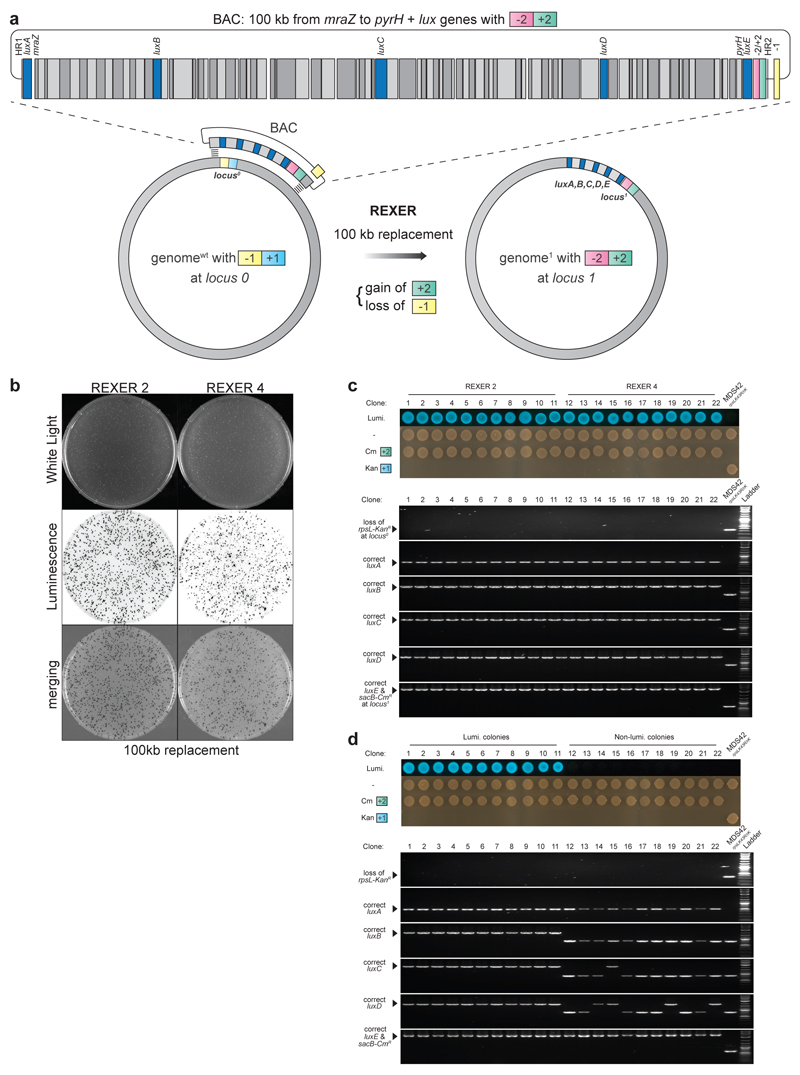
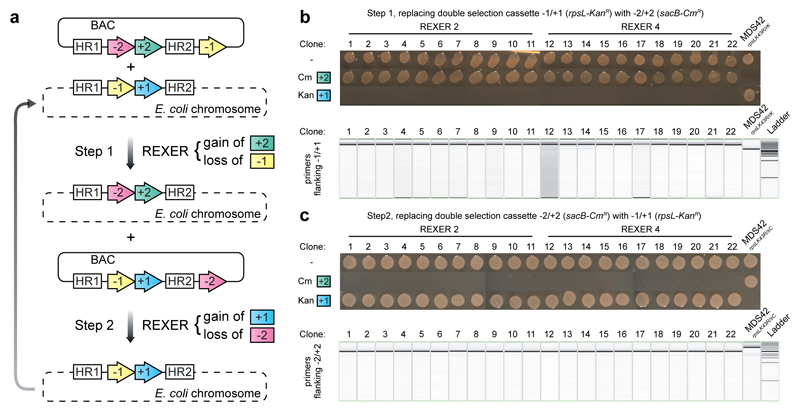
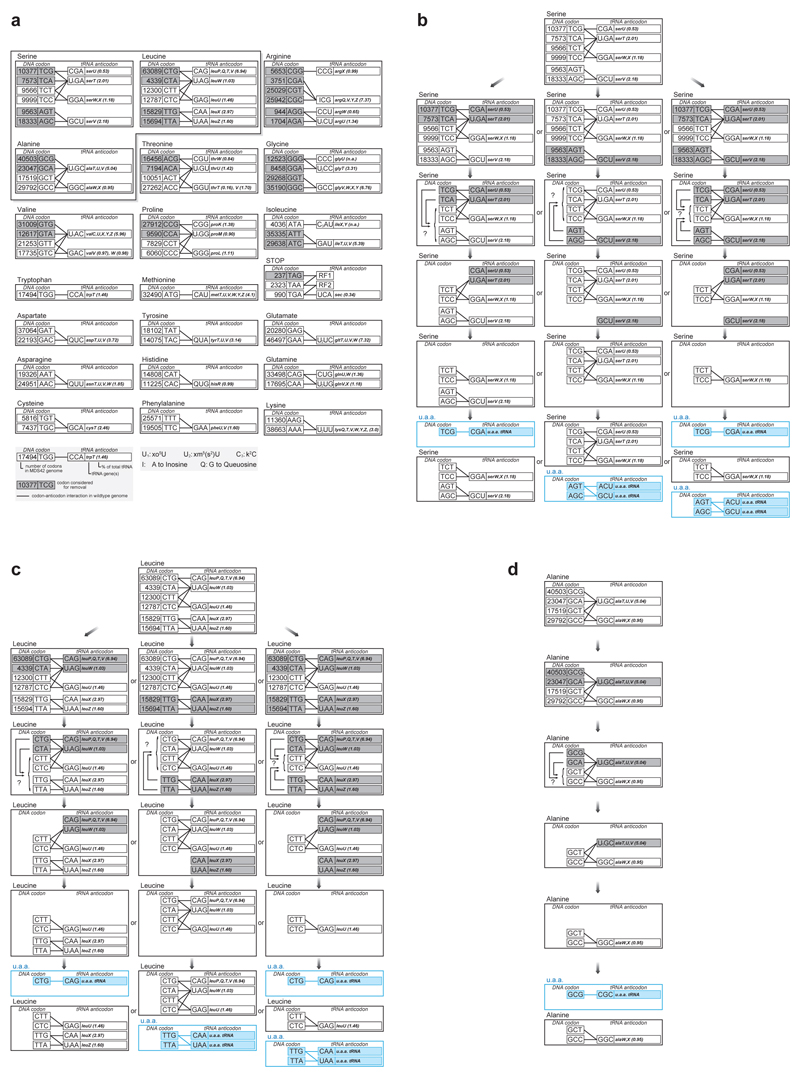
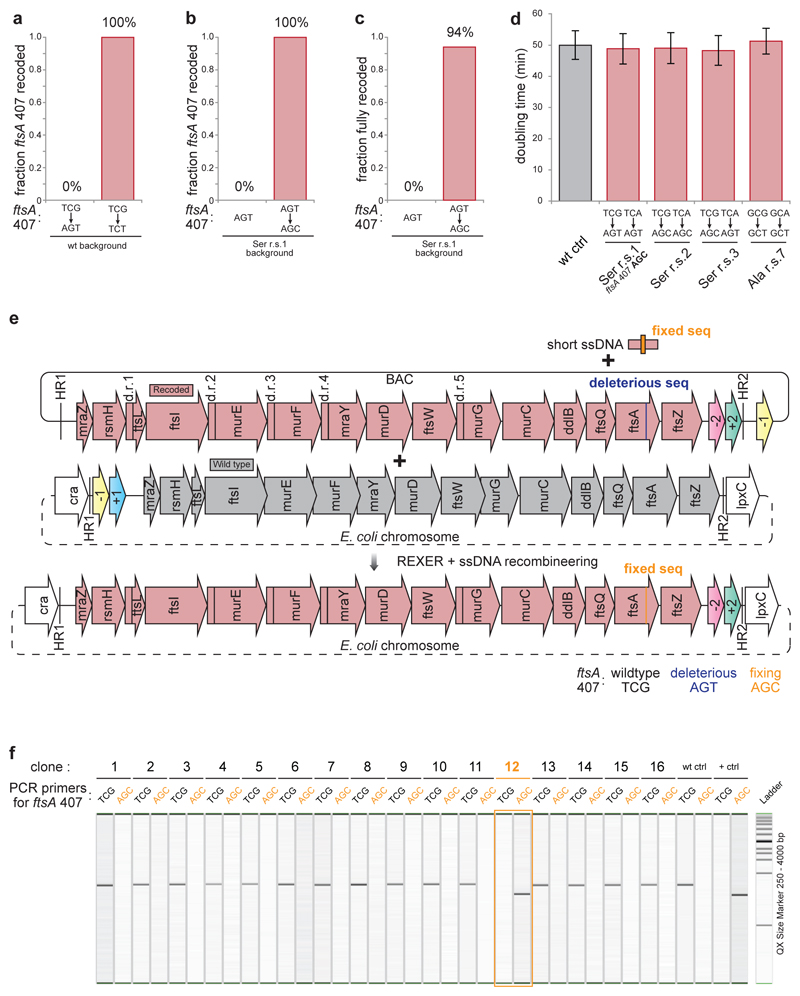
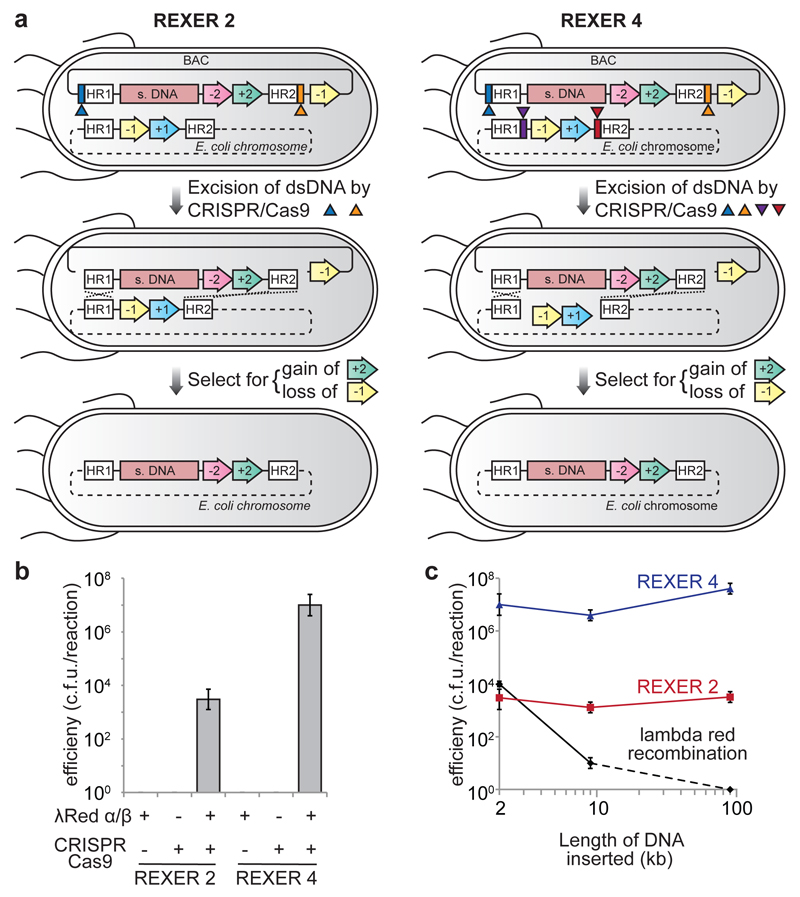
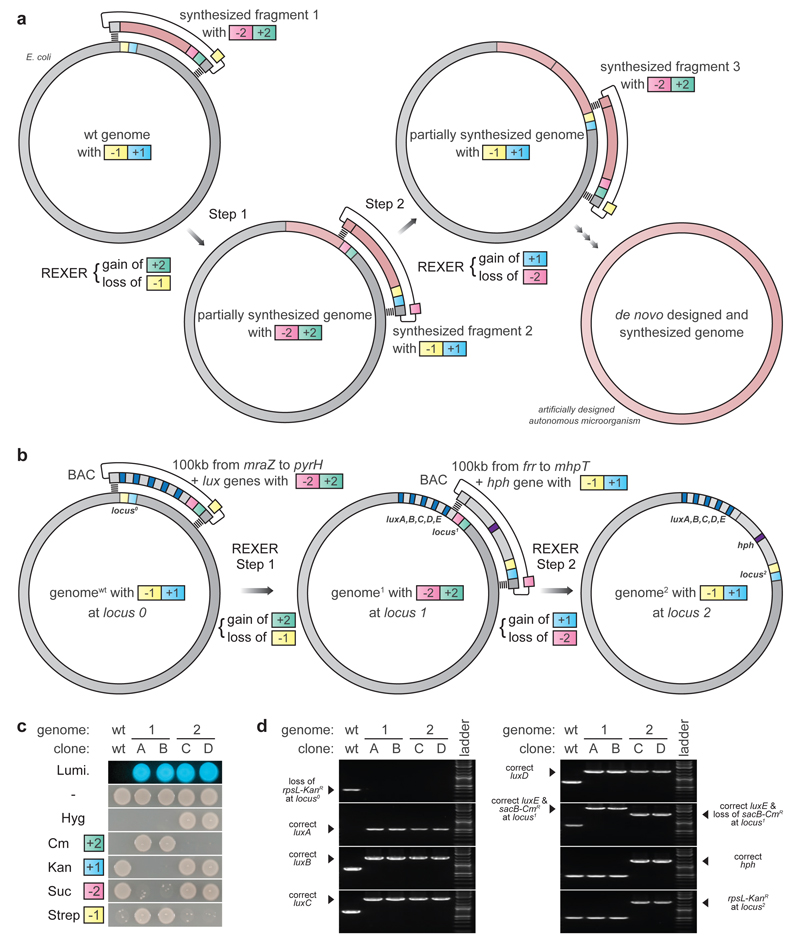
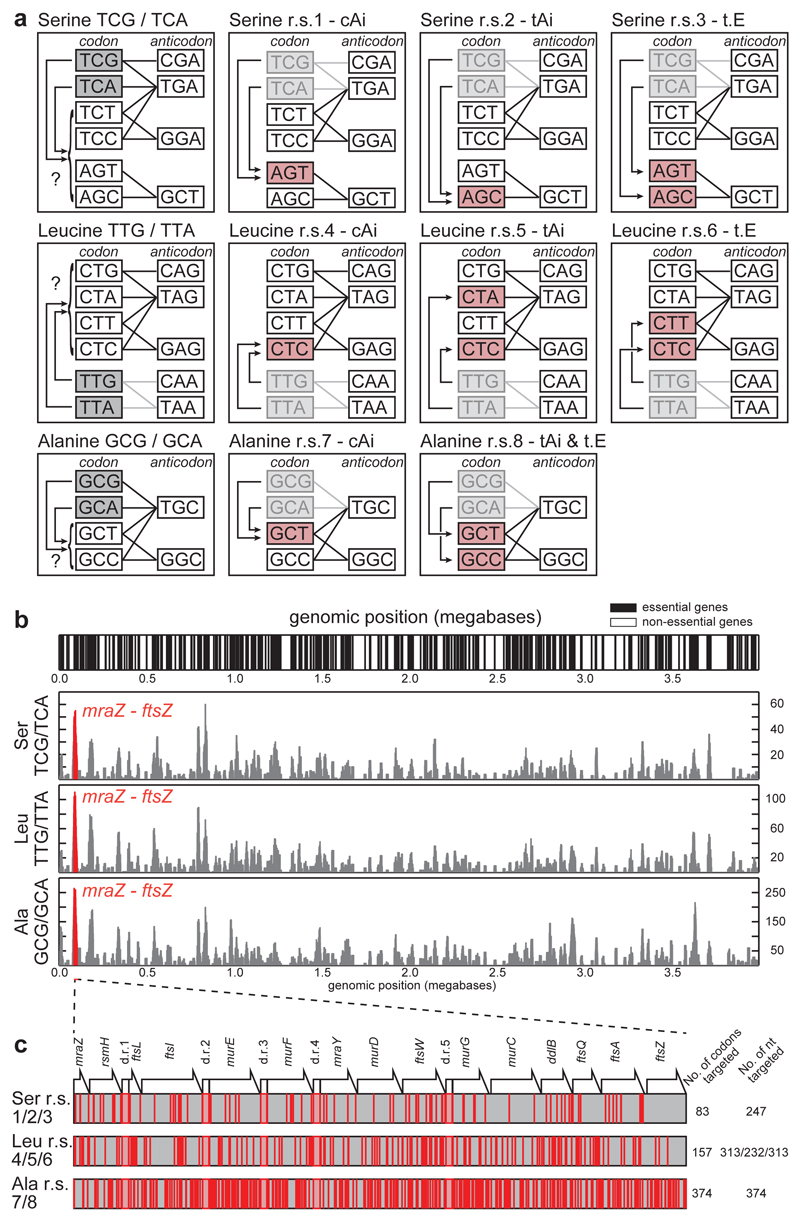
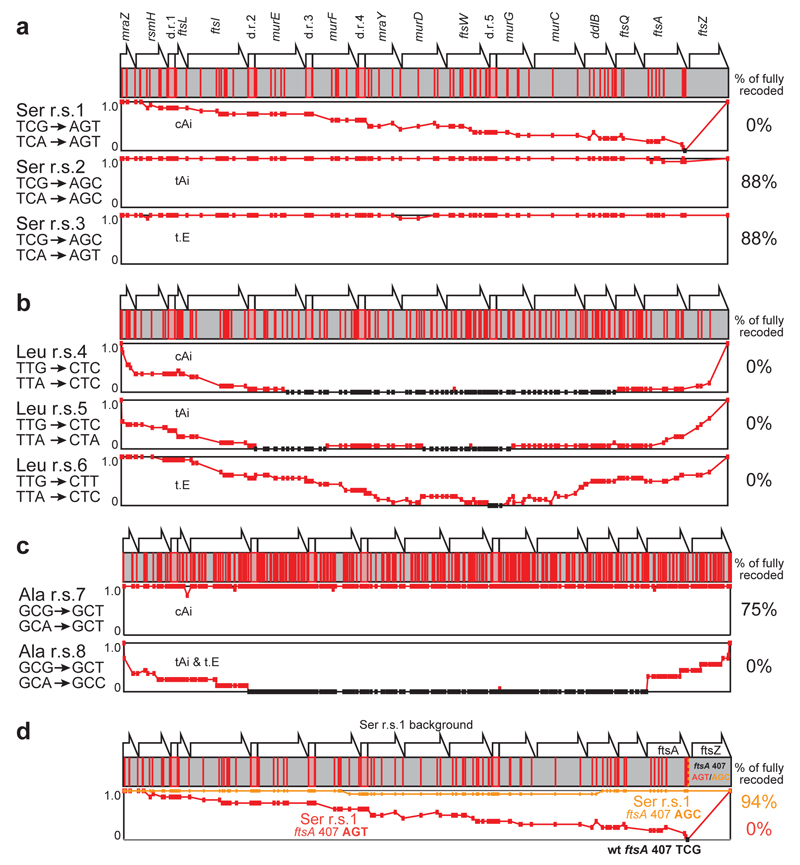
Synthetic recoding of genomes, to remove targeted sense codons, may facilitate the encoded cellular synthesis of unnatural polymers by orthogonal translation systems. However, our limited understanding of allowed synonymous codon substitutions, and the absence of methods that enable the stepwise replacement of the Escherichia coli genome with long synthetic DNA and provide feedback on allowed and disallowed design features in synthetic genomes, have restricted progress towards this goal. Here we endow E. coli with a system for efficient, programmable replacement of genomic DNA with long (>100-kb) synthetic DNA, through the in vivo excision of double-stranded DNA from an episomal replicon by CRISPR/Cas9, coupled to lambda-red-mediated recombination and simultaneous positive and negative selection. We iterate the approach, providing a basis for stepwise whole-genome replacement. We attempt systematic recoding in an essential operon using eight synonymous recoding schemes. Each scheme systematically replaces target codons with defined synonyms and is compatible with codon reassignment. Our results define allowed and disallowed synonymous recoding schemes, and enable the identification and repair of recoding at idiosyncratic positions in the genome.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


Comment in
-
Hacking rules for E. coli.Nat Biotechnol. 2016 Dec 7;34(12):1249. doi: 10.1038/nbt.3744. Nat Biotechnol. 2016. PMID: 27926721 No abstract available.
Similar articles
-
Total synthesis of Escherichia coli with a recoded genome.Nature. 2019 May;569(7757):514-518. doi: 10.1038/s41586-019-1192-5. Epub 2019 May 15. Nature. 2019. PMID: 31092918 Free PMC article.
-
Synthetic genome recoding: new genetic codes for new features.Curr Genet. 2018 Apr;64(2):327-333. doi: 10.1007/s00294-017-0754-z. Epub 2017 Oct 5. Curr Genet. 2018. PMID: 28983660 Free PMC article. Review.
-
Recoded organisms engineered to depend on synthetic amino acids.Nature. 2015 Feb 5;518(7537):89-93. doi: 10.1038/nature14095. Epub 2015 Jan 21. Nature. 2015. PMID: 25607356 Free PMC article.
-
Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality.Proc Natl Acad Sci U S A. 2019 Apr 16;116(16):8070-8079. doi: 10.1073/pnas.1818259116. Epub 2019 Apr 1. Proc Natl Acad Sci U S A. 2019. PMID: 30936302 Free PMC article.
-
Synonymous genome recoding: a tool to explore microbial biology and new therapeutic strategies.Nucleic Acids Res. 2019 Nov 18;47(20):10506-10519. doi: 10.1093/nar/gkz831. Nucleic Acids Res. 2019. PMID: 31584076 Free PMC article. Review.
Cited by
-
Engineered Proteins and Materials Utilizing Residue-Specific Noncanonical Amino Acid Incorporation.Chem Rev. 2024 Aug 14;124(15):9113-9135. doi: 10.1021/acs.chemrev.3c00855. Epub 2024 Jul 15. Chem Rev. 2024. PMID: 39008623 Free PMC article. Review.
-
DNA-assisted selective electrofusion (DASE) of Escherichia coli and giant lipid vesicles.Nanoscale. 2022 Oct 6;14(38):14255-14267. doi: 10.1039/d2nr03105a. Nanoscale. 2022. PMID: 36129323 Free PMC article.
-
Using a quadruplet codon to expand the genetic code of an animal.Nucleic Acids Res. 2022 May 20;50(9):4801-4812. doi: 10.1093/nar/gkab1168. Nucleic Acids Res. 2022. PMID: 34882769 Free PMC article.
-
Editing the microbiome the CRISPR way.Philos Trans R Soc Lond B Biol Sci. 2019 May 13;374(1772):20180103. doi: 10.1098/rstb.2018.0103. Philos Trans R Soc Lond B Biol Sci. 2019. PMID: 30905295 Free PMC article. Review.
-
Simultaneous Multiplex Genome Engineering via Accelerated Natural Transformation in Bacillus subtilis.Front Microbiol. 2021 Aug 17;12:714449. doi: 10.3389/fmicb.2021.714449. eCollection 2021. Front Microbiol. 2021. PMID: 34484154 Free PMC article.
References
-
- Cello J, Paul AV, Wimmer E. Chemical synthesis of poliovirus cDNA: generation of infectious virus in the absence of natural template. Science. 2002;297:1016–1018. - PubMed
-
- Gibson DG, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. - PubMed
-
- Gibson DG, et al. Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science. 2010;329:52–56. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
