

IGSA: Individual Gene Sets Analysis, including Enrichment and Clustering
- PMID: 27764138
- PMCID: PMC5072653
- DOI: 10.1371/journal.pone.0164542
IGSA: Individual Gene Sets Analysis, including Enrichment and Clustering
Abstract
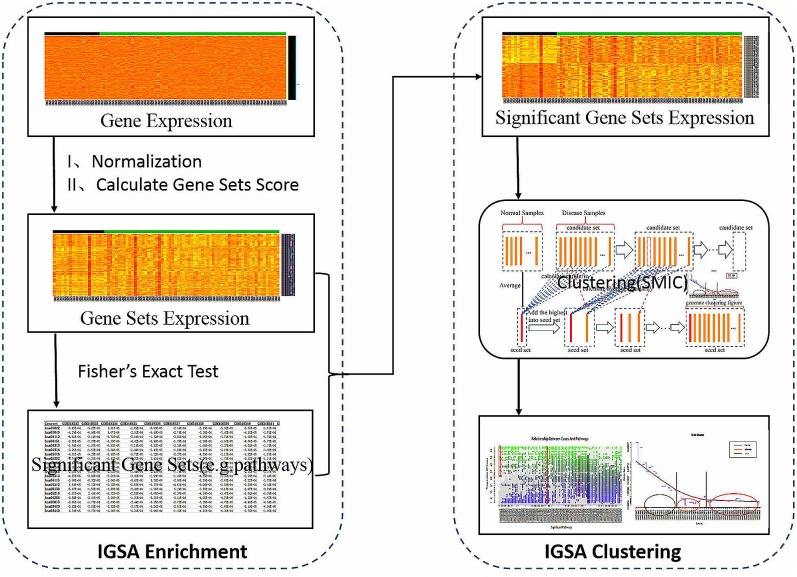
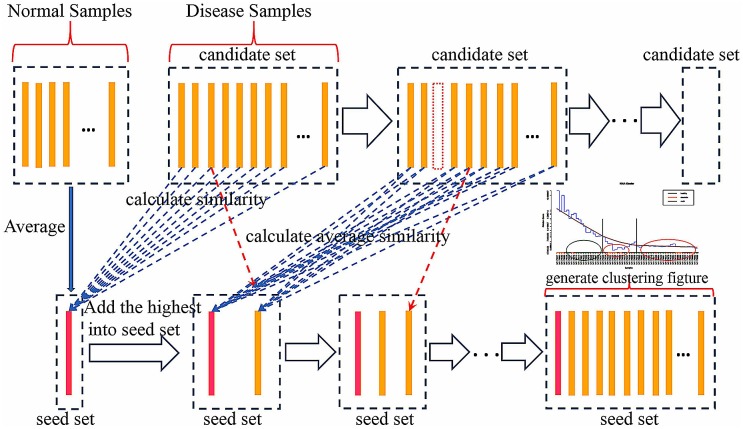
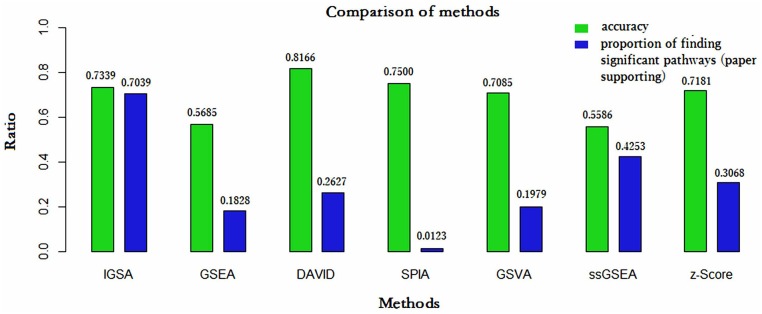
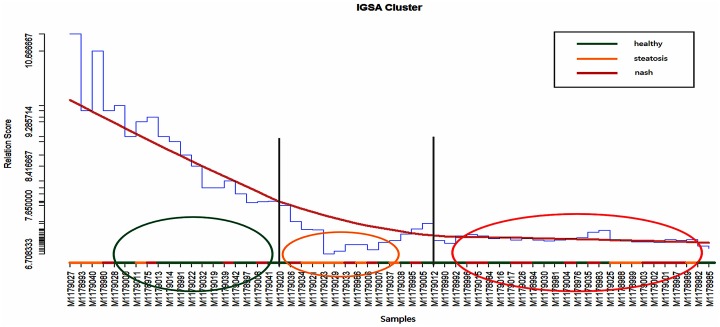
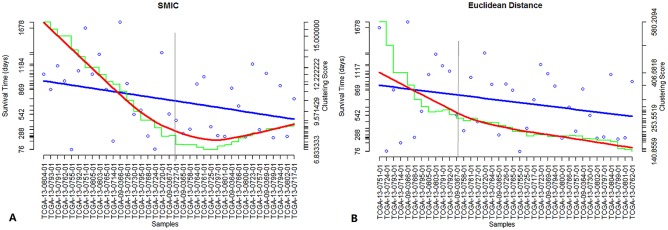
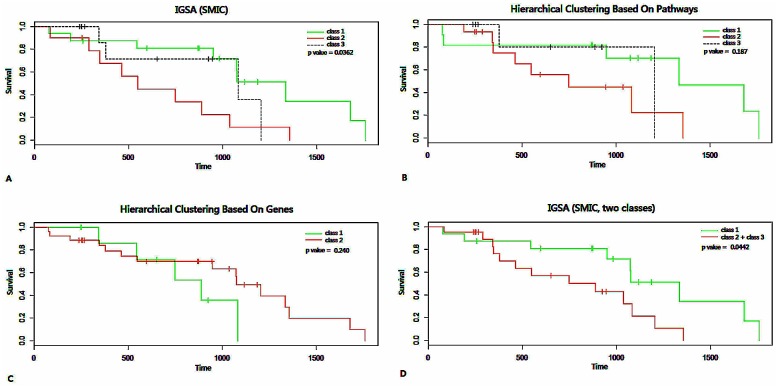
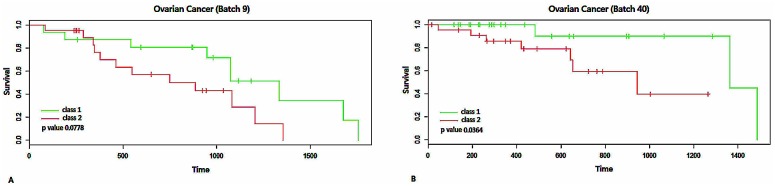
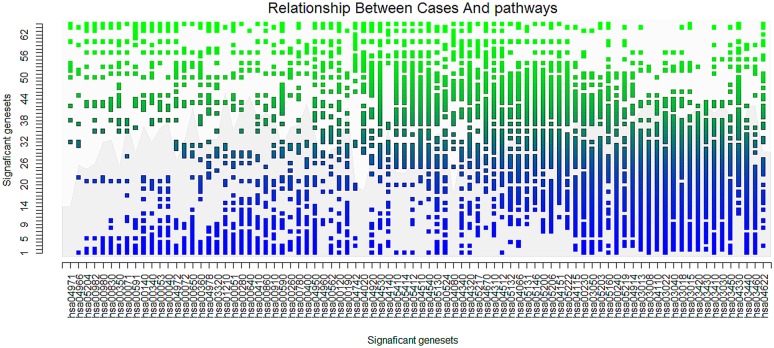
Analysis of gene sets has been widely applied in various high-throughput biological studies. One weakness in the traditional methods is that they neglect the heterogeneity of genes expressions in samples which may lead to the omission of some specific and important gene sets. It is also difficult for them to reflect the severities of disease and provide expression profiles of gene sets for individuals. We developed an application software called IGSA that leverages a powerful analytical capacity in gene sets enrichment and samples clustering. IGSA calculates gene sets expression scores for each sample and takes an accumulating clustering strategy to let the samples gather into the set according to the progress of disease from mild to severe. We focus on gastric, pancreatic and ovarian cancer data sets for the performance of IGSA. We also compared the results of IGSA in KEGG pathways enrichment with David, GSEA, SPIA, ssGSEA and analyzed the results of IGSA clustering and different similarity measurement methods. Notably, IGSA is proved to be more sensitive and specific in finding significant pathways, and can indicate related changes in pathways with the severity of disease. In addition, IGSA provides with significant gene sets profile for each sample.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Gene expression analysis in clear cell renal cell carcinoma using gene set enrichment analysis for biostatistical management.BJU Int. 2011 Jul;108(2 Pt 2):E29-35. doi: 10.1111/j.1464-410X.2010.09794.x. Epub 2011 Mar 16. BJU Int. 2011. PMID: 21435154
-
Detecting discordance enrichment among a series of two-sample genome-wide expression data sets.BMC Genomics. 2017 Jan 25;18(Suppl 1):1050. doi: 10.1186/s12864-016-3265-2. BMC Genomics. 2017. PMID: 28198679 Free PMC article.
-
Pathway enrichment analysis approach based on topological structure and updated annotation of pathway.Brief Bioinform. 2019 Jan 18;20(1):168-177. doi: 10.1093/bib/bbx091. Brief Bioinform. 2019. PMID: 28968630
-
Bioinformatics analysis with graph-based clustering to detect gastric cancer-related pathways.Genet Mol Res. 2012 Sep 26;11(3):3497-504. doi: 10.4238/2012.September.26.5. Genet Mol Res. 2012. PMID: 23079843
-
Gene set enrichment analysis: performance evaluation and usage guidelines.Brief Bioinform. 2012 May;13(3):281-91. doi: 10.1093/bib/bbr049. Epub 2011 Sep 7. Brief Bioinform. 2012. PMID: 21900207 Free PMC article. Review.
Cited by
-
Prognostic risk analysis related to radioresistance genes in colorectal cancer.Front Oncol. 2023 Jan 18;12:1100481. doi: 10.3389/fonc.2022.1100481. eCollection 2022. Front Oncol. 2023. PMID: 36741692 Free PMC article.
-
Comprehensive Analysis of Tumor Microenvironment Identified Prognostic Immune-Related Gene Signature in Ovarian Cancer.Front Genet. 2021 Feb 10;12:616073. doi: 10.3389/fgene.2021.616073. eCollection 2021. Front Genet. 2021. PMID: 33679883 Free PMC article.
-
Integrating RNA sequencing into neuro-oncology practice.Transl Res. 2017 Nov;189:93-104. doi: 10.1016/j.trsl.2017.06.013. Epub 2017 Jul 8. Transl Res. 2017. PMID: 28746860 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
