

Structural quality of unrefined models in protein docking
- PMID: 27756103
- PMCID: PMC5167671
- DOI: 10.1002/prot.25188
Structural quality of unrefined models in protein docking
Abstract
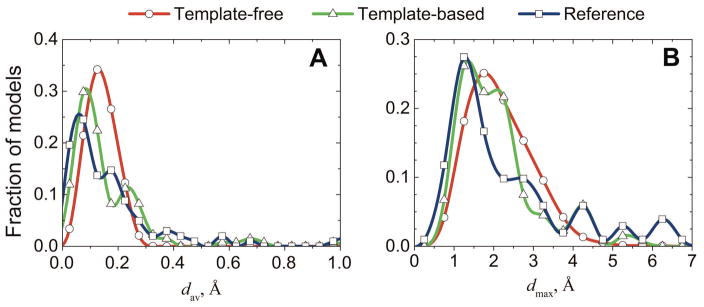
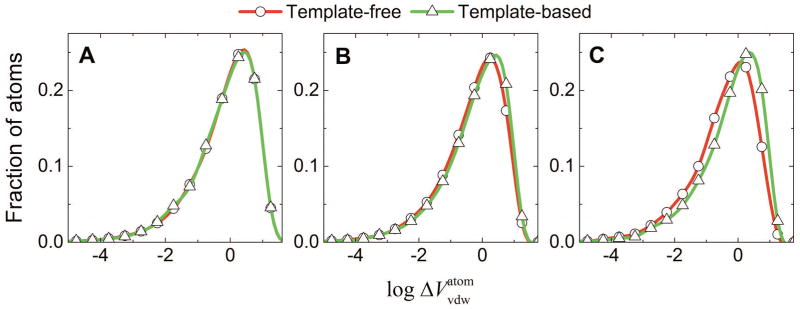
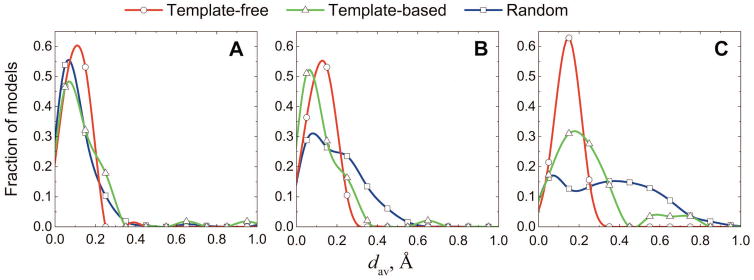
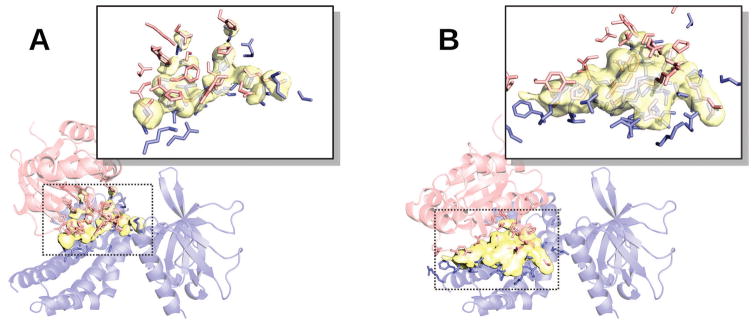
Structural characterization of protein-protein interactions is essential for understanding life processes at the molecular level. However, only a fraction of protein interactions have experimentally resolved structures. Thus, reliable computational methods for structural modeling of protein interactions (protein docking) are important for generating such structures and understanding the principles of protein recognition. Template-based docking techniques that utilize structural similarity between target protein-protein interaction and cocrystallized protein-protein complexes (templates) are gaining popularity due to generally higher reliability than that of the template-free docking. However, the template-based approach lacks explicit penalties for intermolecular penetration, as opposed to the typical free docking where such penalty is inherent due to the shape complementarity paradigm. Thus, template-based docking models are commonly assumed to require special treatment to remove large structural penetrations. In this study, we compared clashes in the template-based and free docking of the same proteins, with crystallographically determined and modeled structures. The results show that for the less accurate protein models, free docking produces fewer clashes than the template-based approach. However, contrary to the common expectation, in acceptable and better quality docking models of unbound crystallographically determined proteins, the clashes in the template-based docking are comparable to those in the free docking, due to the overall higher quality of the template-based docking predictions. This suggests that the free docking refinement protocols can in principle be applied to the template-based docking predictions as well. Proteins 2016; 85:39-45. © 2016 Wiley Periodicals, Inc.
Keywords: interactome; model refinement; protein modeling; protein recognition; steric clash; structure prediction.
© 2016 Wiley Periodicals, Inc.
Figures
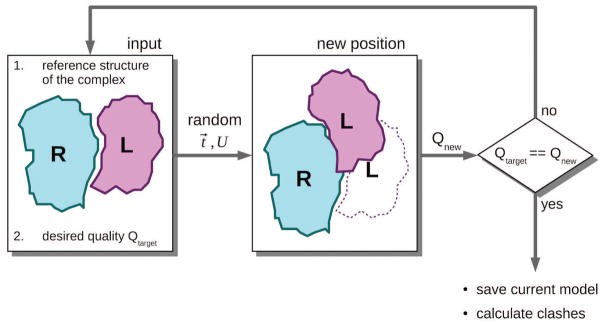
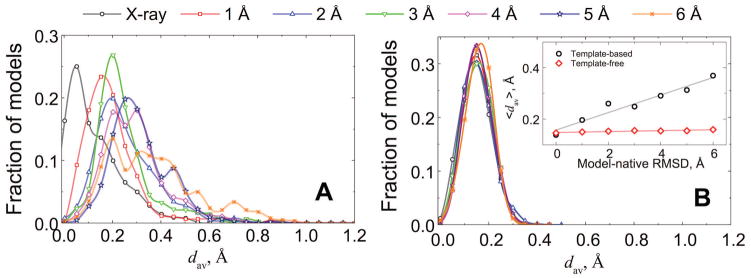
Similar articles
-
Modeling complexes of modeled proteins.Proteins. 2017 Mar;85(3):470-478. doi: 10.1002/prot.25183. Epub 2016 Oct 24. Proteins. 2017. PMID: 27701777 Free PMC article.
-
How to choose templates for modeling of protein complexes: Insights from benchmarking template-based docking.Proteins. 2020 Aug;88(8):1070-1081. doi: 10.1002/prot.25875. Epub 2020 Feb 7. Proteins. 2020. PMID: 31994759 Free PMC article.
-
Addressing recent docking challenges: A hybrid strategy to integrate template-based and free protein-protein docking.Proteins. 2017 Mar;85(3):497-512. doi: 10.1002/prot.25234. Epub 2017 Jan 24. Proteins. 2017. PMID: 28026062
-
What method to use for protein-protein docking?Curr Opin Struct Biol. 2019 Apr;55:1-7. doi: 10.1016/j.sbi.2018.12.010. Epub 2019 Feb 1. Curr Opin Struct Biol. 2019. PMID: 30711743 Free PMC article. Review.
-
Low-resolution structural modeling of protein interactome.Curr Opin Struct Biol. 2013 Apr;23(2):198-205. doi: 10.1016/j.sbi.2012.12.003. Epub 2013 Jan 5. Curr Opin Struct Biol. 2013. PMID: 23294579 Free PMC article. Review.
Cited by
-
GWYRE: A Resource for Mapping Variants onto Experimental and Modeled Structures of Human Protein Complexes.J Mol Biol. 2022 Jun 15;434(11):167608. doi: 10.1016/j.jmb.2022.167608. Epub 2022 Apr 27. J Mol Biol. 2022. PMID: 35662458 Free PMC article.
-
Prediction of Host-Pathogen Interactions for Helicobacter pylori by Interface Mimicry and Implications to Gastric Cancer.J Mol Biol. 2017 Dec 8;429(24):3925-3941. doi: 10.1016/j.jmb.2017.10.023. Epub 2017 Oct 26. J Mol Biol. 2017. PMID: 29106933 Free PMC article.
-
Oncoviruses Can Drive Cancer by Rewiring Signaling Pathways Through Interface Mimicry.Front Oncol. 2019 Nov 15;9:1236. doi: 10.3389/fonc.2019.01236. eCollection 2019. Front Oncol. 2019. PMID: 31803618 Free PMC article.
-
Modeling CAPRI targets 110-120 by template-based and free docking using contact potential and combined scoring function.Proteins. 2018 Mar;86 Suppl 1(Suppl 1):302-310. doi: 10.1002/prot.25380. Epub 2017 Sep 28. Proteins. 2018. PMID: 28905425 Free PMC article.
References
-
- Moal IH, Moretti R, Baker D, Fernandez-Recio J. Scoring functions for protein–protein interactions. Curr Opin Struct Biol. 2013;23:862–867. - PubMed
-
- Hunjan J, Tovchigrechko A, Gao Y, Vakser IA. The size of the intermolecular energy funnel in protein-protein interactions. Proteins. 2008;72:344–352. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
