

Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer
- PMID: 27748765
- PMCID: PMC5245767
- DOI: 10.1038/onc.2016.382
Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer
Abstract
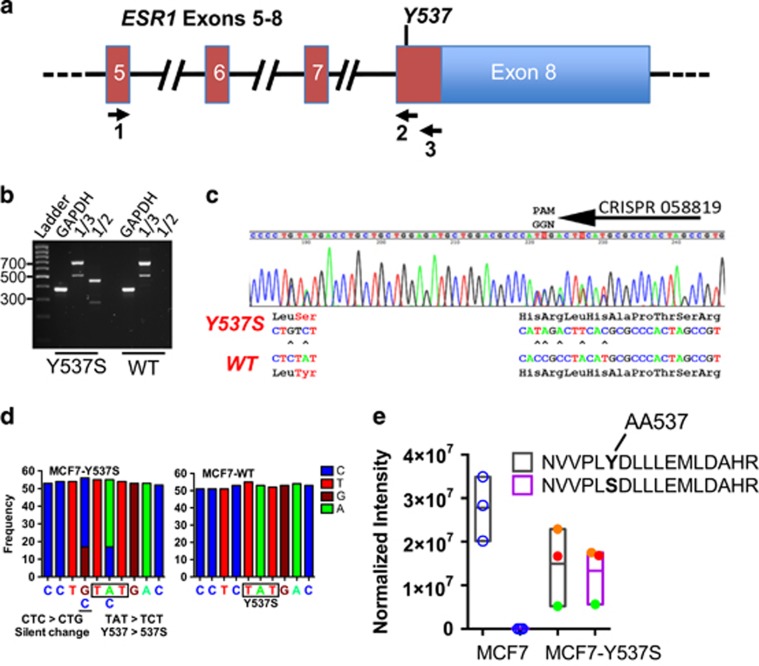
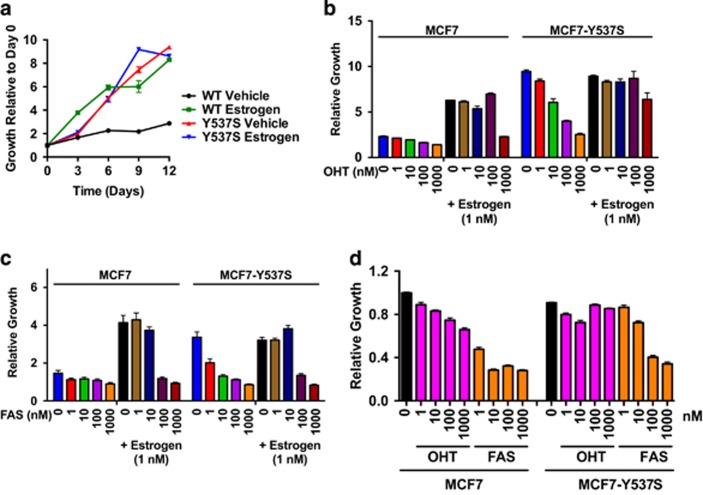
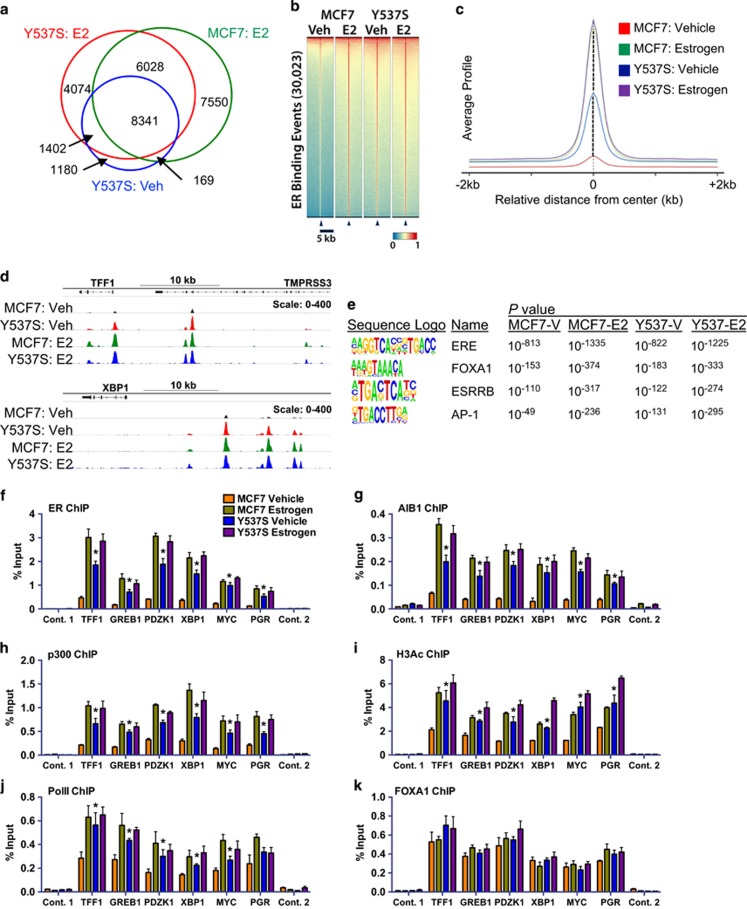
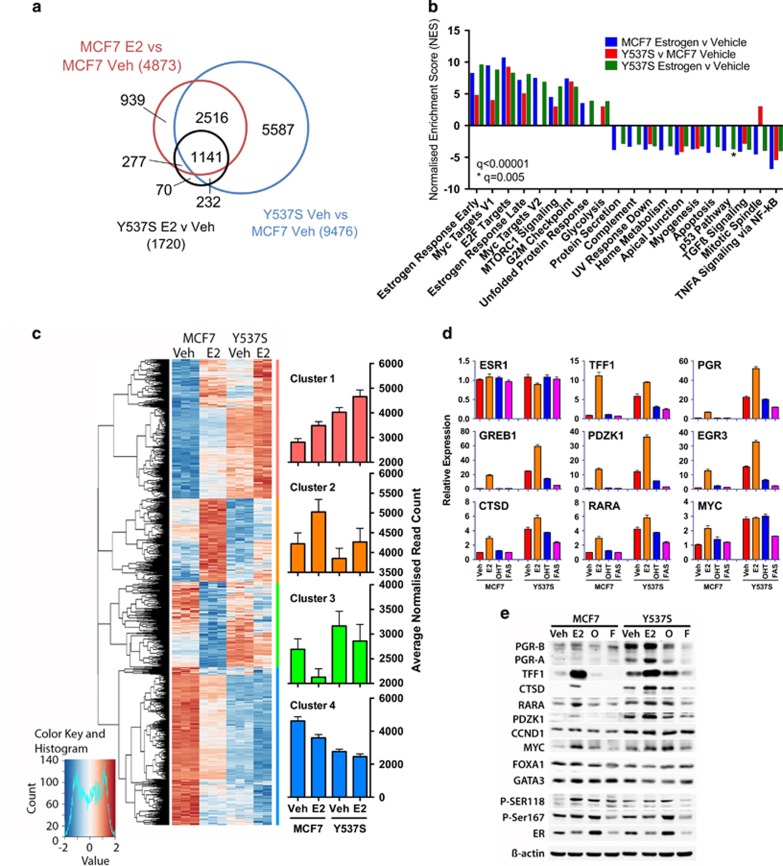
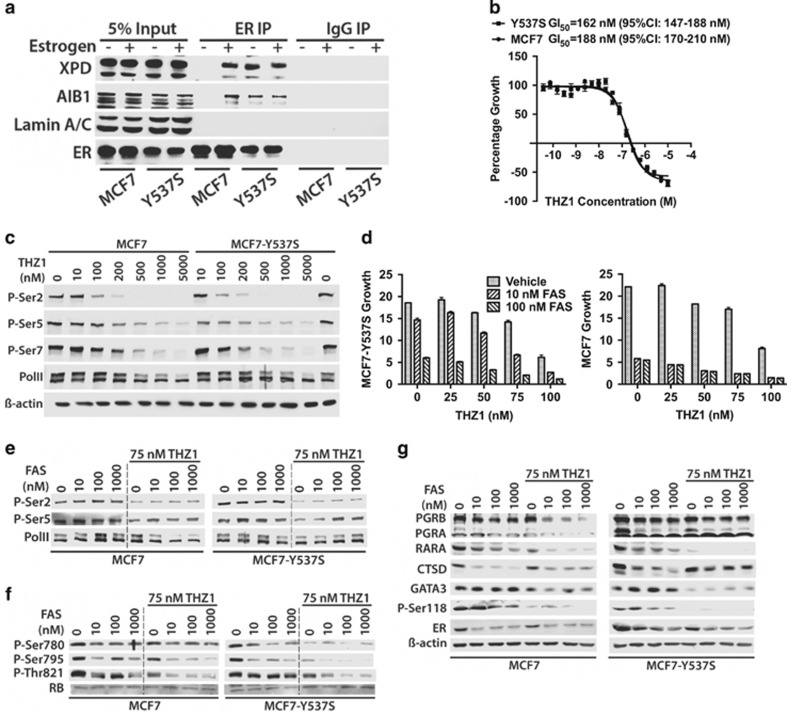
Drugs that inhibit estrogen receptor-α (ER) activity have been highly successful in treating and reducing breast cancer progression in ER-positive disease. However, resistance to these therapies presents a major clinical problem. Recent genetic studies have shown that mutations in the ER gene are found in >20% of tumours that progress on endocrine therapies. Remarkably, the great majority of these mutations localize to just a few amino acids within or near the critical helix 12 region of the ER hormone binding domain, where they are likely to be single allele mutations. Understanding how these mutations impact on ER function is a prerequisite for identifying methods to treat breast cancer patients featuring such mutations. Towards this end, we used CRISPR-Cas9 genome editing to make a single allele knock-in of the most commonly mutated amino acid residue, tyrosine 537, in the estrogen-responsive MCF7 breast cancer cell line. Genomic analyses using RNA-seq and ER ChIP-seq demonstrated that the Y537S mutation promotes constitutive ER activity globally, resulting in estrogen-independent growth. MCF7-Y537S cells were resistant to the anti-estrogen tamoxifen and fulvestrant. Further, we show that the basal transcription factor TFIIH is constitutively recruited by ER-Y537S, resulting in ligand-independent phosphorylation of Serine 118 (Ser118) by the TFIIH kinase, cyclin-dependent kinase (CDK)7. The CDK7 inhibitor, THZ1 prevented Ser118 phosphorylation and inhibited growth of MCF7-Y537S cells. These studies confirm the functional importance of ER mutations in endocrine resistance, demonstrate the utility of knock-in mutational models for investigating alternative therapeutic approaches and highlight CDK7 inhibition as a potential therapy for endocrine-resistant breast cancer mediated by ER mutations.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Targeted degradation of activating estrogen receptor α ligand-binding domain mutations in human breast cancer.Breast Cancer Res Treat. 2020 Apr;180(3):611-622. doi: 10.1007/s10549-020-05564-y. Epub 2020 Feb 17. Breast Cancer Res Treat. 2020. PMID: 32067153
-
Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models.Breast Cancer Res. 2017 May 23;19(1):60. doi: 10.1186/s13058-017-0851-4. Breast Cancer Res. 2017. PMID: 28535794 Free PMC article.
-
Longitudinal Molecular Imaging of Progesterone Receptor Reveals Early Differential Response to Endocrine Therapy in Breast Cancer with an Activating ESR1 Mutation.J Nucl Med. 2021 Apr;62(4):500-506. doi: 10.2967/jnumed.120.249508. Epub 2020 Aug 28. J Nucl Med. 2021. PMID: 32859700 Free PMC article.
-
ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer.Breast Cancer Res. 2021 Aug 15;23(1):85. doi: 10.1186/s13058-021-01462-3. Breast Cancer Res. 2021. PMID: 34392831 Free PMC article. Review.
-
The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials.Biochim Biophys Acta Rev Cancer. 2019 Dec;1872(2):188315. doi: 10.1016/j.bbcan.2019.188315. Epub 2019 Oct 21. Biochim Biophys Acta Rev Cancer. 2019. PMID: 31647985 Review.
Cited by
-
Clinically relevant CHK1 inhibitors abrogate wild-type and Y537S mutant ERα expression and proliferation in luminal primary and metastatic breast cancer cells.J Exp Clin Cancer Res. 2022 Apr 13;41(1):141. doi: 10.1186/s13046-022-02360-y. J Exp Clin Cancer Res. 2022. PMID: 35418303 Free PMC article.
-
HNRNPA2B1 regulates tamoxifen- and fulvestrant-sensitivity and hallmarks of endocrine resistance in breast cancer cells.Cancer Lett. 2021 Oct 10;518:152-168. doi: 10.1016/j.canlet.2021.07.015. Epub 2021 Jul 14. Cancer Lett. 2021. PMID: 34273466 Free PMC article.
-
Epigenetic mechanisms in breast cancer therapy and resistance.Nat Commun. 2021 Mar 19;12(1):1786. doi: 10.1038/s41467-021-22024-3. Nat Commun. 2021. PMID: 33741974 Free PMC article. Review.
-
Systematic Characterization of Recurrent Genomic Alterations in Cyclin-Dependent Kinases Reveals Potential Therapeutic Strategies for Cancer Treatment.Cell Rep. 2020 Jul 14;32(2):107884. doi: 10.1016/j.celrep.2020.107884. Cell Rep. 2020. PMID: 32668240 Free PMC article.
-
ABC-transporter upregulation mediates resistance to the CDK7 inhibitors THZ1 and ICEC0942.Oncogene. 2020 Jan;39(3):651-663. doi: 10.1038/s41388-019-1008-y. Epub 2019 Sep 17. Oncogene. 2020. PMID: 31530935 Free PMC article.
References
-
- Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol 2010; 11: 1135–1141. - PubMed
-
- Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med 1998; 339: 1609–1618. - PubMed
-
- Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2002; 2: 101–112. - PubMed
-
- Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med 2011; 62: 217–232. - PubMed
-
- Johnston SR, Dowsett M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer 2003; 3: 821–831. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
