

Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles
- PMID: 27741508
- PMCID: PMC5356914
- DOI: 10.18632/oncotarget.12554
Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles
Abstract
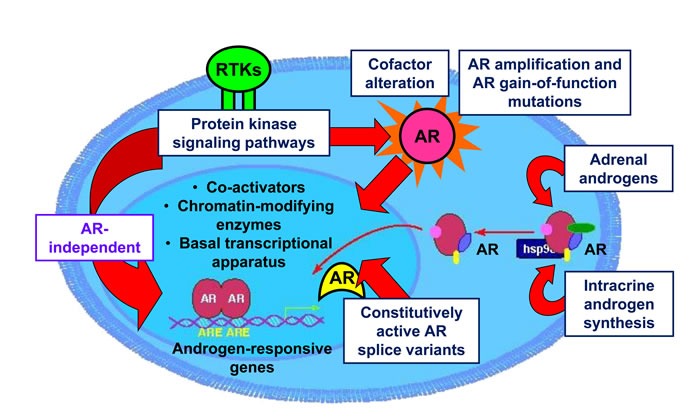
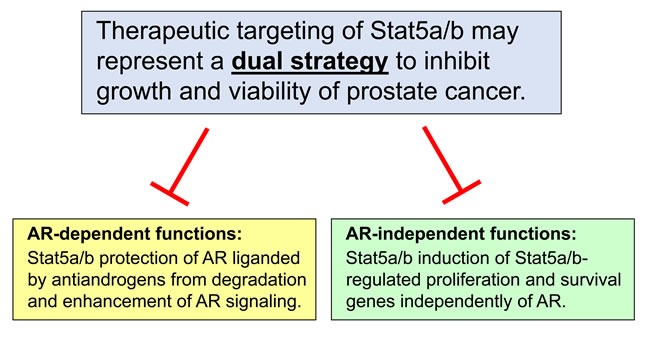
Despite aggressive treatment for localized cancer, prostate cancer (PC) remains a leading cause of cancer-related death for American men due to a subset of patients progressing to lethal and incurable metastatic castrate-resistant prostate cancer (CRPC). Organ-confined PC is treated by surgery or radiation with or without androgen deprivation therapy (ADT), while options for locally advanced and disseminated PC include radiation combined with ADT, or systemic treatments including chemotherapy. Progression to CRPC results from failure of ADT, which targets the androgen receptor (AR) signaling axis and inhibits AR-driven proliferation and survival pathways. The exact mechanisms underlying the transition from androgen-dependent PC to CRPC remain incompletely understood. Reactivation of AR has been shown to occur in CRPC despite depletion of circulating androgens by ADT. At the same time, the presence of AR-negative cell populations in CRPC has also been identified. While AR signaling has been proposed as the primary driver of CRPC, AR-independent signaling pathways may represent additional mechanisms underlying CRPC progression. Identification of new therapeutic strategies to target both AR-positive and AR-negative PC cell populations and, thereby, AR-driven as well as non-AR-driven PC cell growth and survival mechanisms would provide a two-pronged approach to eliminate CRPC cells with potential for synthetic lethality. In this review, we provide an overview of AR-dependent and AR-independent molecular mechanisms which drive CRPC, with special emphasis on the role of the Jak2-Stat5a/b signaling pathway in promoting castrate-resistant growth of PC through both AR-dependent and AR-independent mechanisms.
Keywords: Jak2; Stat5a/b; androgen receptor; antiandrogen; castrate-resistant; metastasis; prostate cancer.
Conflict of interest statement
The authors declare no conflict of interests.
Figures

Similar articles
-
Targeting the androgen receptor signaling pathway in advanced prostate cancer.Am J Health Syst Pharm. 2022 Jul 22;79(15):1224-1235. doi: 10.1093/ajhp/zxac105. Am J Health Syst Pharm. 2022. PMID: 35390118 Review.
-
Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis.Oncogene. 2014 May 29;33(22):2815-25. doi: 10.1038/onc.2013.235. Epub 2013 Jun 10. Oncogene. 2014. PMID: 23752196 Free PMC article. Review.
-
Arginine vasopressin receptor 1a is a therapeutic target for castration-resistant prostate cancer.Sci Transl Med. 2019 Jun 26;11(498):eaaw4636. doi: 10.1126/scitranslmed.aaw4636. Sci Transl Med. 2019. PMID: 31243151 Free PMC article.
-
Androgen receptors in hormone-dependent and castration-resistant prostate cancer.Pharmacol Ther. 2013 Dec;140(3):223-38. doi: 10.1016/j.pharmthera.2013.07.003. Epub 2013 Jul 13. Pharmacol Ther. 2013. PMID: 23859952 Review.
-
Targeting the MIF/CXCR7/AKT Signaling Pathway in Castration-Resistant Prostate Cancer.Mol Cancer Res. 2019 Jan;17(1):263-276. doi: 10.1158/1541-7786.MCR-18-0412. Epub 2018 Sep 17. Mol Cancer Res. 2019. PMID: 30224544 Free PMC article.
Cited by
-
Clinico-Pathological Factors and AR-LBD Mutations in Early and Late Castration-Resistant Prostate Cancer.Cancer Manag Res. 2024 Oct 21;16:1509-1516. doi: 10.2147/CMAR.S477439. eCollection 2024. Cancer Manag Res. 2024. PMID: 39464307 Free PMC article.
-
Calcium signalling pathways in prostate cancer initiation and progression.Nat Rev Urol. 2023 Sep;20(9):524-543. doi: 10.1038/s41585-023-00738-x. Epub 2023 Mar 24. Nat Rev Urol. 2023. PMID: 36964408 Review.
-
STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells.Int J Mol Sci. 2019 Apr 12;20(8):1815. doi: 10.3390/ijms20081815. Int J Mol Sci. 2019. PMID: 31013746 Free PMC article.
-
The Key Characteristics of Carcinogens: Relationship to the Hallmarks of Cancer, Relevant Biomarkers, and Assays to Measure Them.Cancer Epidemiol Biomarkers Prev. 2020 Oct;29(10):1887-1903. doi: 10.1158/1055-9965.EPI-19-1346. Epub 2020 Mar 9. Cancer Epidemiol Biomarkers Prev. 2020. PMID: 32152214 Free PMC article.
-
MicroRNA-126 inhibits proliferation and metastasis in prostate cancer via regulation of ADAM9.Oncol Lett. 2018 Jun;15(6):9051-9060. doi: 10.3892/ol.2018.8528. Epub 2018 Apr 18. Oncol Lett. 2018. PMID: 29805636 Free PMC article.
References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. - PubMed
-
- Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65(11):1180–1192. - PubMed
-
- Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, de Bono JS. Prostate cancer. Lancet. 2016;387(10013):70–82. - PubMed
-
- Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513–1520. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
