

ADAM17 substrate release in proximal tubule drives kidney fibrosis
- PMID: 27642633
- PMCID: PMC5026414
- DOI: 10.1172/jci.insight.87023
ADAM17 substrate release in proximal tubule drives kidney fibrosis
Abstract
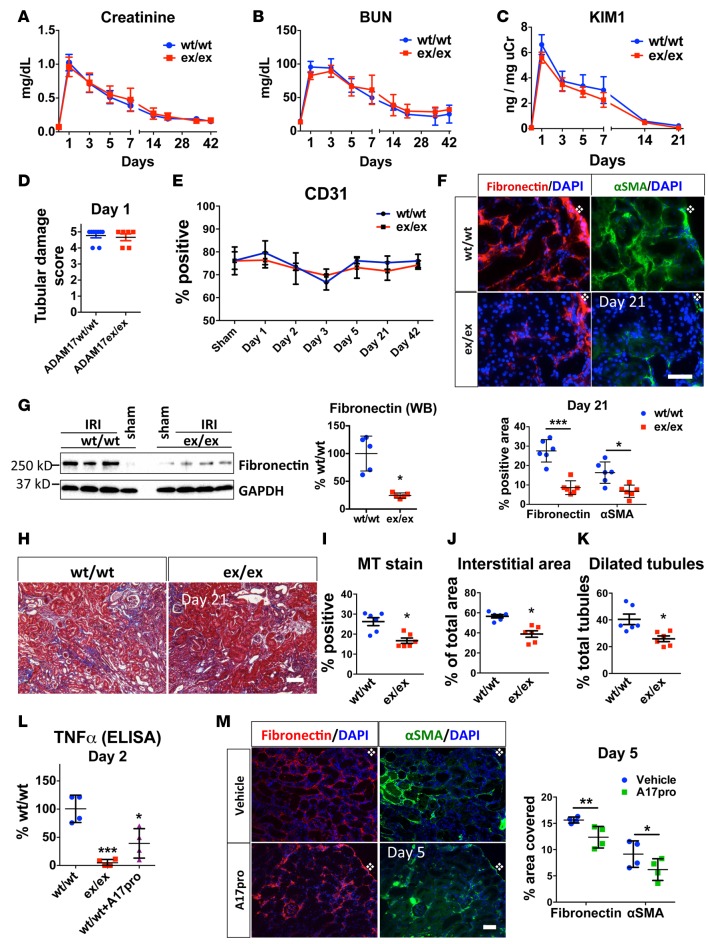
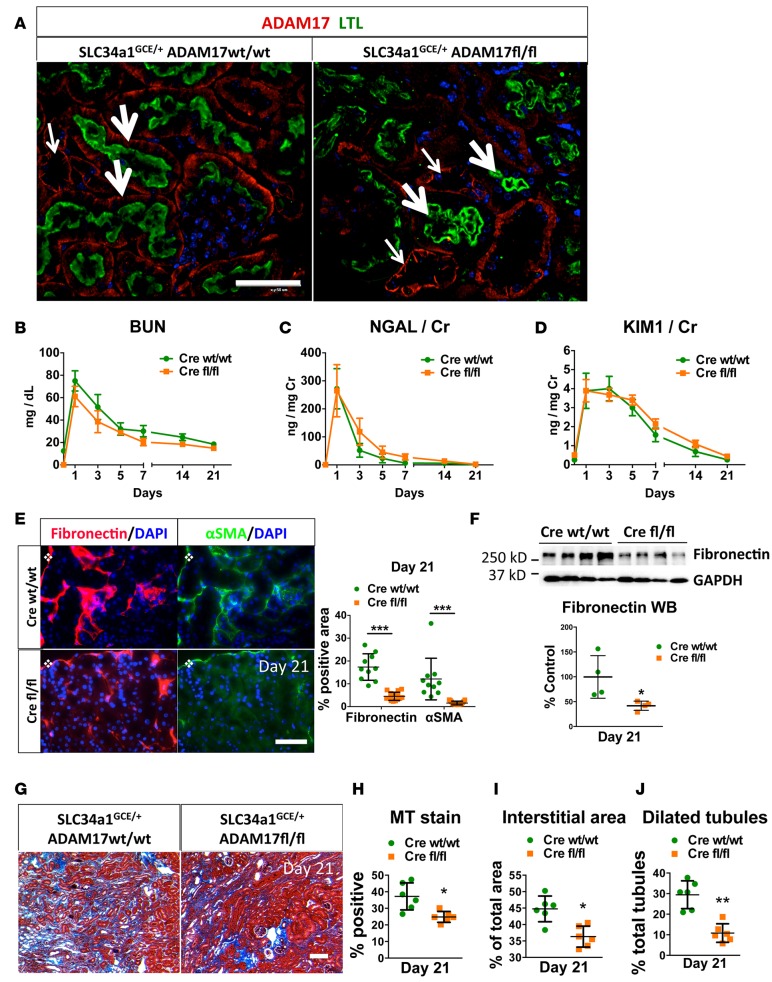
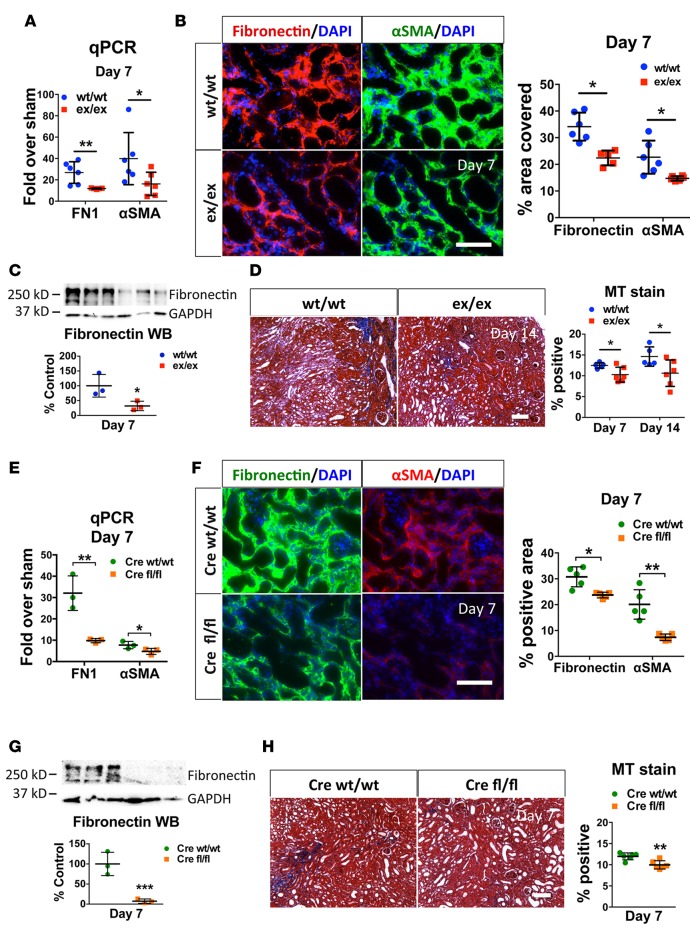
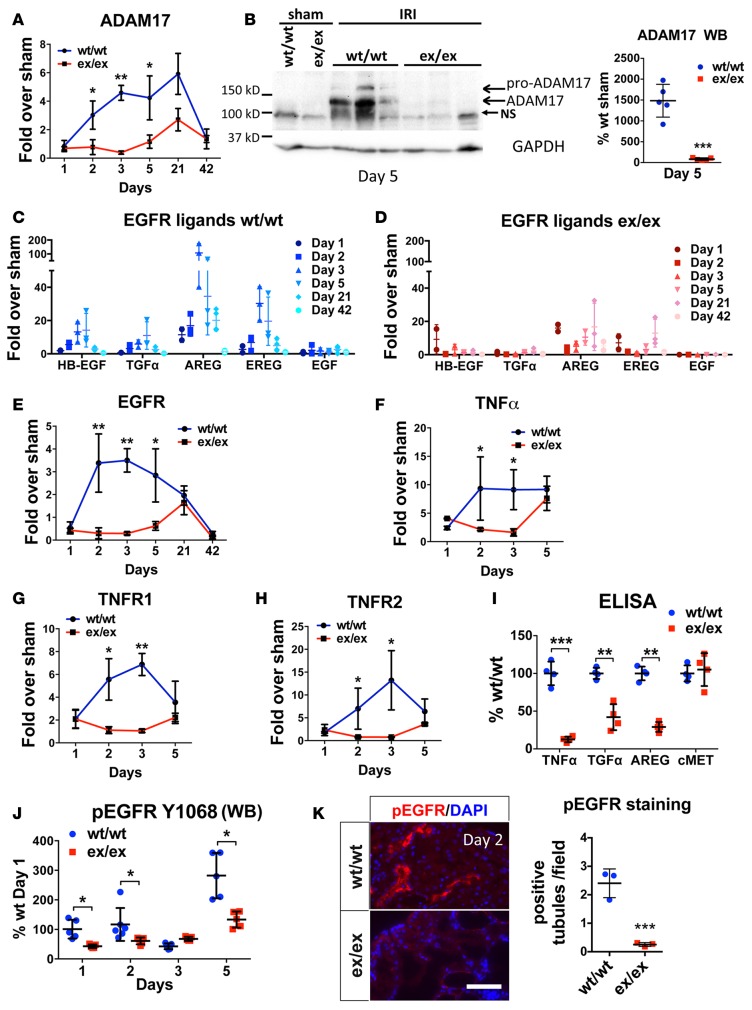
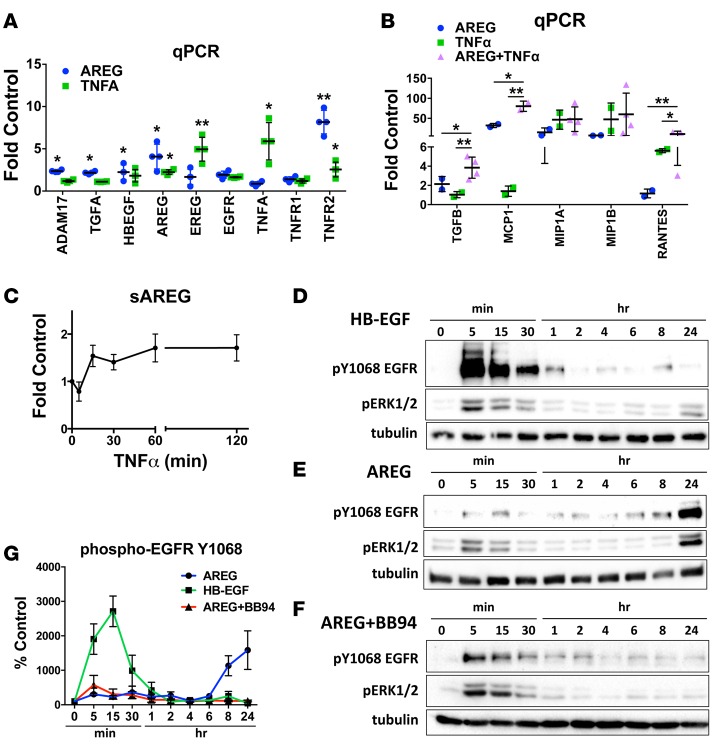
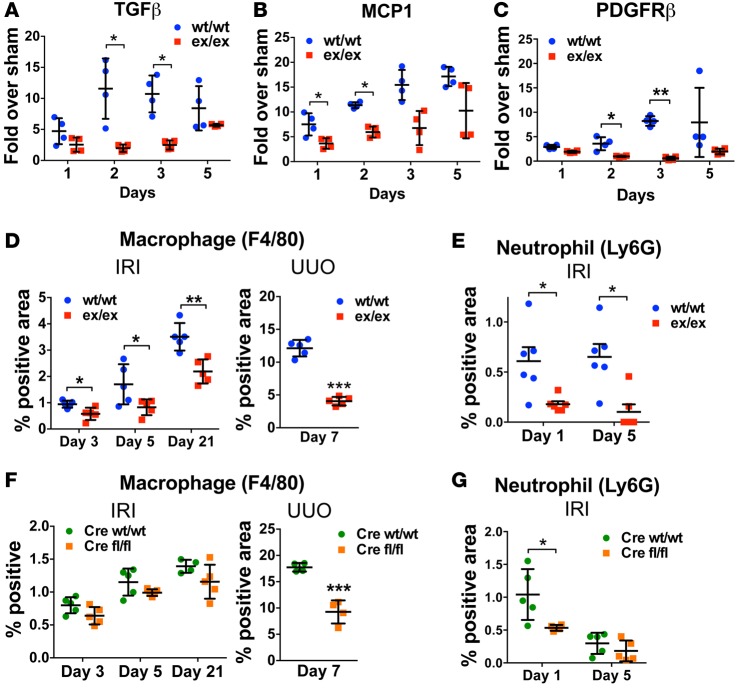
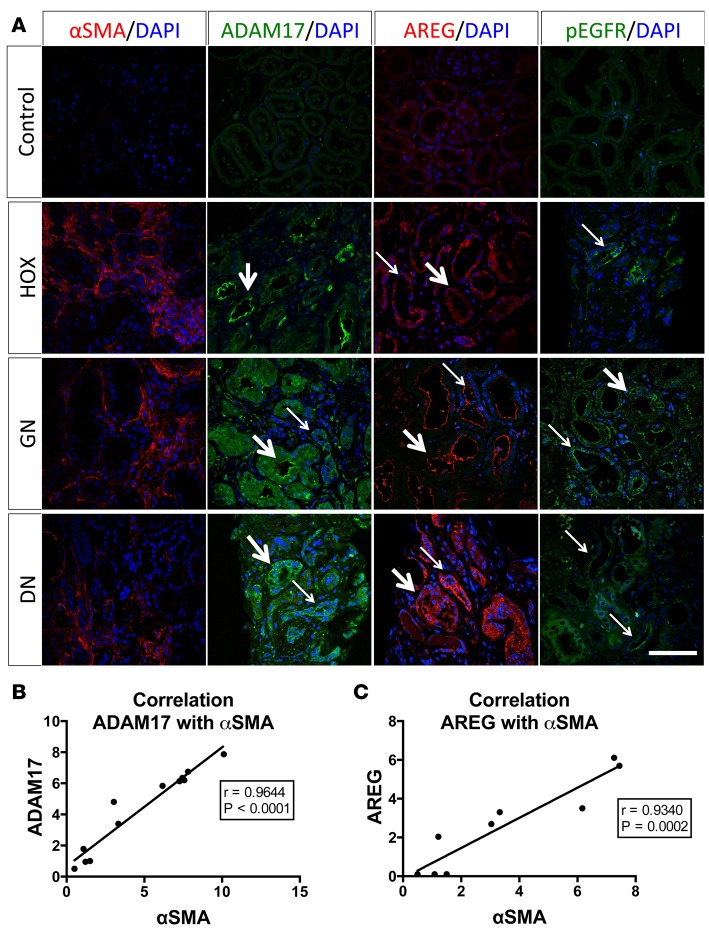
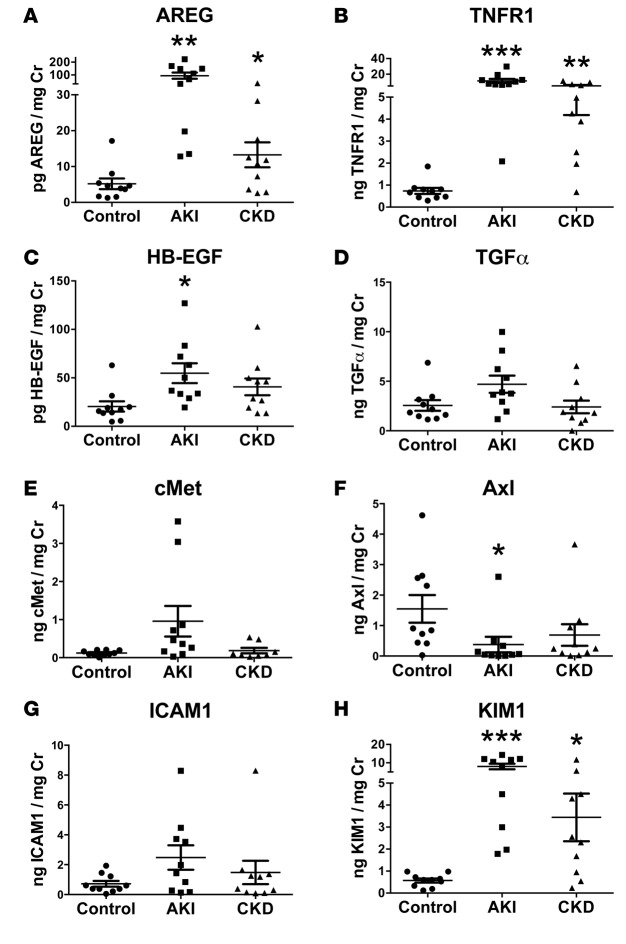
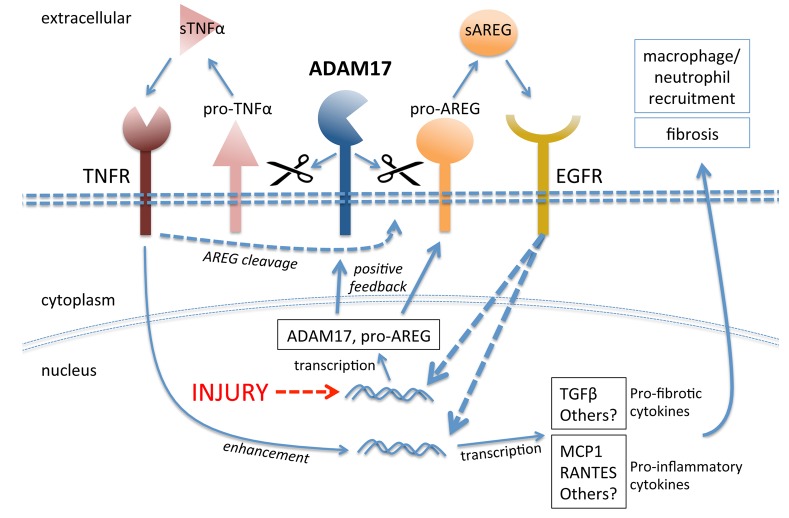
Kidney fibrosis following kidney injury is an unresolved health problem and causes significant morbidity and mortality worldwide. In a study into its molecular mechanism, we identified essential causative features. Acute or chronic kidney injury causes sustained elevation of a disintegrin and metalloprotease 17 (ADAM17); of its cleavage-activated proligand substrates, in particular of pro-TNFα and the EGFR ligand amphiregulin (pro-AREG); and of the substrates' receptors. As a consequence, EGFR is persistently activated and triggers the synthesis and release of proinflammatory and profibrotic factors, resulting in macrophage/neutrophil ingress and fibrosis. ADAM17 hypomorphic mice, specific ADAM17 inhibitor-treated WT mice, or mice with inducible KO of ADAM17 in proximal tubule (Slc34a1-Cre) were significantly protected against these effects. In vitro, in proximal tubule cells, we show that AREG has unique profibrotic actions that are potentiated by TNFα-induced AREG cleavage. In vivo, in acute kidney injury (AKI) and chronic kidney disease (CKD, fibrosis) patients, soluble AREG is indeed highly upregulated in human urine, and both ADAM17 and AREG expression show strong positive correlation with fibrosis markers in related kidney biopsies. Our results indicate that targeting of the ADAM17 pathway represents a therapeutic target for human kidney fibrosis.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures
Similar articles
-
Proximal Tubule-Derived Amphiregulin Amplifies and Integrates Profibrotic EGF Receptor Signals in Kidney Fibrosis.J Am Soc Nephrol. 2019 Dec;30(12):2370-2383. doi: 10.1681/ASN.2019030321. Epub 2019 Nov 1. J Am Soc Nephrol. 2019. PMID: 31676723 Free PMC article.
-
Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity.Am J Physiol Lung Cell Mol Physiol. 2018 Apr 1;314(4):L555-L568. doi: 10.1152/ajplung.00458.2017. Epub 2017 Dec 14. Am J Physiol Lung Cell Mol Physiol. 2018. PMID: 29351448
-
KIM-1-mediated anti-inflammatory activity is preserved by MUC1 induction in the proximal tubule during ischemia-reperfusion injury.Am J Physiol Renal Physiol. 2021 Aug 1;321(2):F135-F148. doi: 10.1152/ajprenal.00127.2021. Epub 2021 Jun 21. Am J Physiol Renal Physiol. 2021. PMID: 34151589 Free PMC article.
-
Role of ADAM17 in kidney disease.Am J Physiol Renal Physiol. 2019 Aug 1;317(2):F333-F342. doi: 10.1152/ajprenal.00625.2018. Epub 2019 May 29. Am J Physiol Renal Physiol. 2019. PMID: 31141400 Review.
-
The EGFR-ADAM17 Axis in Chronic Obstructive Pulmonary Disease and Cystic Fibrosis Lung Pathology.Mediators Inflamm. 2018 Jan 9;2018:1067134. doi: 10.1155/2018/1067134. eCollection 2018. Mediators Inflamm. 2018. PMID: 29540993 Free PMC article. Review.
Cited by
-
Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion.JCI Insight. 2017 Sep 21;2(18):e94716. doi: 10.1172/jci.insight.94716. eCollection 2017 Sep 21. JCI Insight. 2017. PMID: 28931758 Free PMC article.
-
Renal ADAM10 and 17: Their Physiological and Medical Meanings.Front Cell Dev Biol. 2018 Nov 6;6:153. doi: 10.3389/fcell.2018.00153. eCollection 2018. Front Cell Dev Biol. 2018. PMID: 30460232 Free PMC article.
-
Blocking CHOP-dependent TXNIP shuttling to mitochondria attenuates albuminuria and mitigates kidney injury in nephrotic syndrome.Proc Natl Acad Sci U S A. 2022 Aug 30;119(35):e2116505119. doi: 10.1073/pnas.2116505119. Epub 2022 Aug 22. Proc Natl Acad Sci U S A. 2022. PMID: 35994650 Free PMC article.
-
iRhom2 promotes lupus nephritis through TNF-α and EGFR signaling.J Clin Invest. 2018 Apr 2;128(4):1397-1412. doi: 10.1172/JCI97650. Epub 2018 Mar 5. J Clin Invest. 2018. PMID: 29369823 Free PMC article.
-
Proximal tubule LPA1 and LPA2 receptors use divergent signaling pathways to additively increase profibrotic cytokine secretion.Am J Physiol Renal Physiol. 2021 Mar 1;320(3):F359-F374. doi: 10.1152/ajprenal.00494.2020. Epub 2021 Jan 11. Am J Physiol Renal Physiol. 2021. PMID: 33427061 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
