

Changes in cardiac Nav1.5 expression, function, and acetylation by pan-histone deacetylase inhibitors
- PMID: 27638876
- PMCID: PMC5130491
- DOI: 10.1152/ajpheart.00156.2016
Changes in cardiac Nav1.5 expression, function, and acetylation by pan-histone deacetylase inhibitors
Abstract
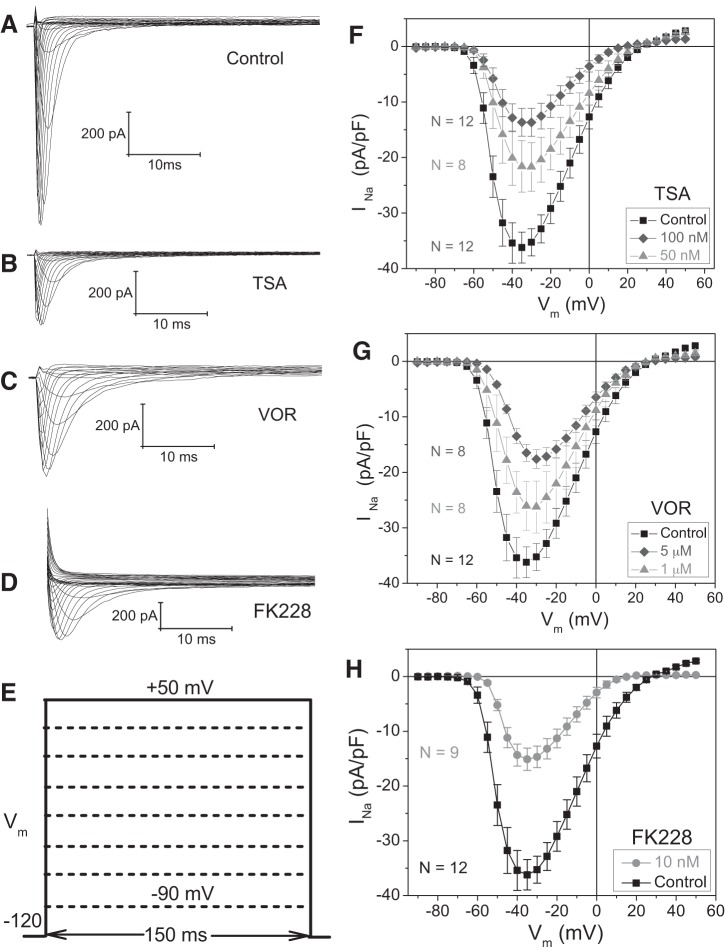
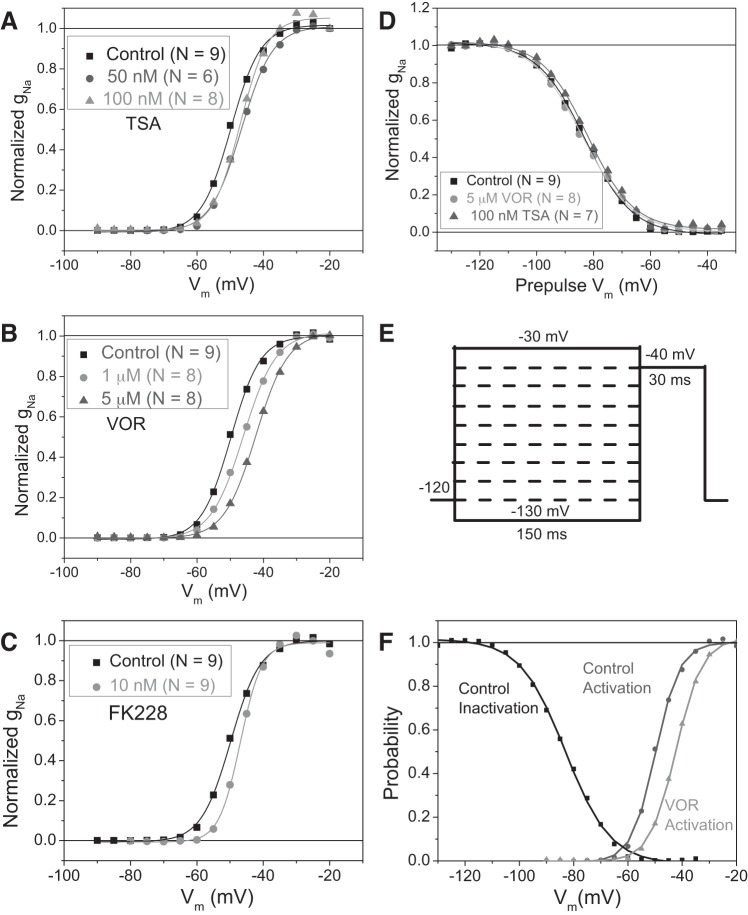
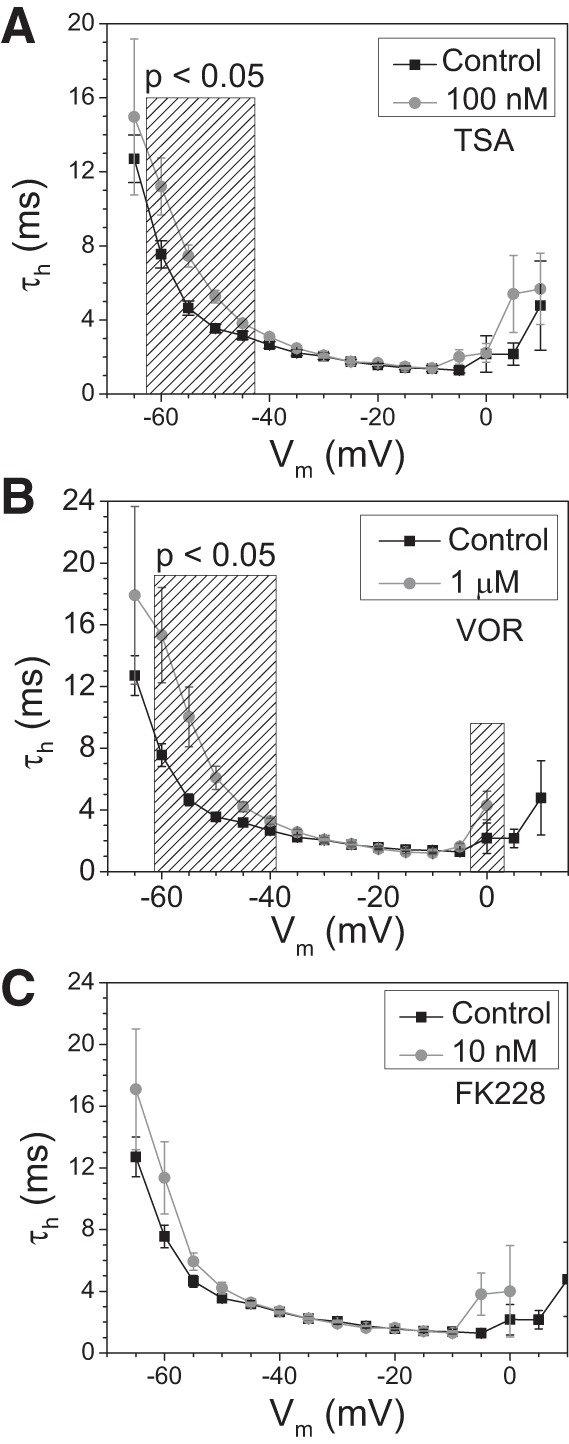
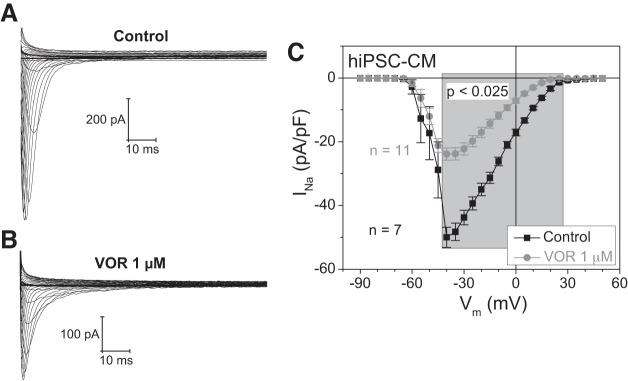
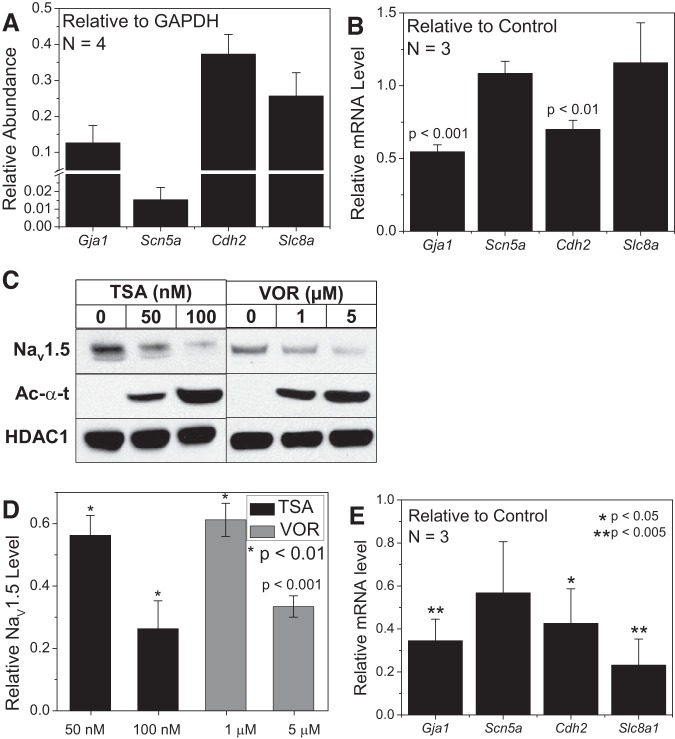
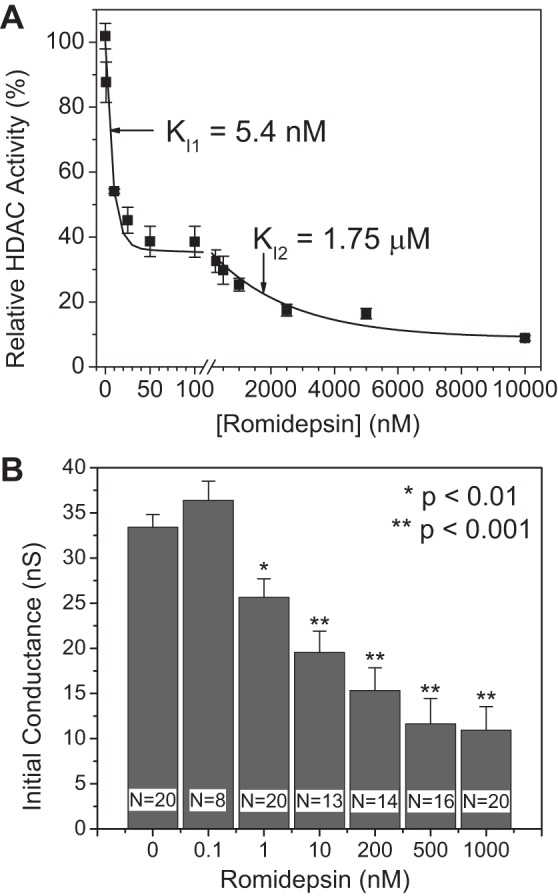
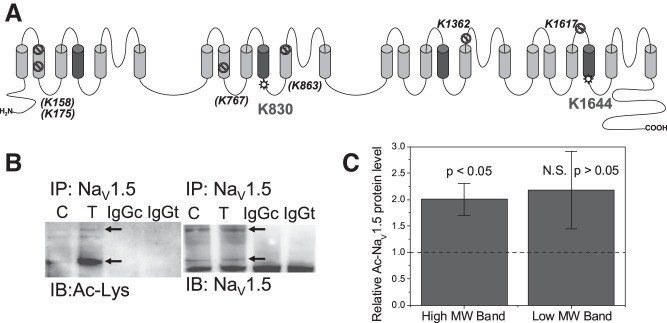
Histone deacetylase (HDAC) inhibitors are small molecule anticancer therapeutics that exhibit limiting cardiotoxicities including QT interval prolongation and life-threatening cardiac arrhythmias. Because the molecular mechanisms for HDAC inhibitor-induced cardiotoxicity are poorly understood, we performed whole cell patch voltage-clamp experiments to measure cardiac sodium currents (INa) from wild-type neonatal mouse ventricular or human-induced pluripotent stem cell-derived cardiomyocytes treated with trichostatin A (TSA), vorinostat (VOR), or romidepsin (FK228). All three pan-HDAC inhibitors dose dependently decreased peak INa density and shifted the voltage activation curve 3- to 8-mV positive. Increases in late INa were not observed despite a moderate slowing of the inactivation rate at low activating potentials (<-40 mV). Scn5a mRNA levels were not significantly altered but NaV1.5 protein levels were significantly reduced. Immunoprecipitation with anti-NaV1.5 and Western blotting with anti-acetyl-lysine antibodies indicated that NaV1.5 acetylation is increased in vivo after HDAC inhibition. FK228 inhibited total cardiac HDAC activity with two apparent IC50s of 5 nM and 1.75 μM, consistent with previous findings with TSA and VOR. FK228 also decreased ventricular gap junction conductance (gj), again consistent with previous findings. We conclude that pan-HDAC inhibition reduces cardiac INa density and NaV1.5 protein levels without affecting late INa amplitude and, thus, probably does not contribute to the reported QT interval prolongation and arrhythmias associated with pan-HDAC inhibitor therapies. Conversely, reductions in gj may enhance the occurrence of triggered activity by limiting electrotonic inhibition and, combined with reduced INa, slow myocardial conduction and increase vulnerability to reentrant arrhythmias.
Keywords: gap junctions; romidepsin; sodium current; trichostatin A; vorinostat.
Copyright © 2016 the American Physiological Society.
Figures
Similar articles
-
Differences in Functional Expression of Connexin43 and NaV1.5 by Pan- and Class-Selective Histone Deacetylase Inhibition in Heart.Int J Mol Sci. 2018 Aug 4;19(8):2288. doi: 10.3390/ijms19082288. Int J Mol Sci. 2018. PMID: 30081552 Free PMC article.
-
Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/- and human SCN5A-1795insD+/- iPSC-derived cardiomyocytes.Cardiovasc Res. 2017 Jun 1;113(7):829-838. doi: 10.1093/cvr/cvx077. Cardiovasc Res. 2017. PMID: 28430892
-
Sirtuin 1 regulates cardiac electrical activity by deacetylating the cardiac sodium channel.Nat Med. 2017 Mar;23(3):361-367. doi: 10.1038/nm.4284. Epub 2017 Feb 13. Nat Med. 2017. PMID: 28191886 Free PMC article.
-
Dysfunctional Nav1.5 channels due to SCN5A mutations.Exp Biol Med (Maywood). 2018 Jun;243(10):852-863. doi: 10.1177/1535370218777972. Epub 2018 May 27. Exp Biol Med (Maywood). 2018. PMID: 29806494 Free PMC article. Review.
-
Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: ionic and non-ionic mechanisms.Cardiovasc Res. 2020 Jul 15;116(9):1557-1570. doi: 10.1093/cvr/cvaa082. Cardiovasc Res. 2020. PMID: 32251506 Free PMC article. Review.
Cited by
-
Pharmacogenomics in drug-induced cardiotoxicity: Current status and the future.Front Cardiovasc Med. 2022 Oct 13;9:966261. doi: 10.3389/fcvm.2022.966261. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36312261 Free PMC article. Review.
-
hiPSCs in cardio-oncology: deciphering the genomics.Cardiovasc Res. 2019 Apr 15;115(5):935-948. doi: 10.1093/cvr/cvz018. Cardiovasc Res. 2019. PMID: 30689737 Free PMC article. Review.
-
Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology.Cells. 2022 Jan 7;11(2):200. doi: 10.3390/cells11020200. Cells. 2022. PMID: 35053315 Free PMC article. Review.
-
From HDAC to Voltage-Gated Ion Channels: What's Next? The Long Road of Antiepileptic Drugs Repositioning in Cancer.Cancers (Basel). 2022 Sep 10;14(18):4401. doi: 10.3390/cancers14184401. Cancers (Basel). 2022. PMID: 36139561 Free PMC article. Review.
-
Differential regulation of KCa 2.1 (KCNN1) K+ channel expression by histone deacetylases in atrial fibrillation with concomitant heart failure.Physiol Rep. 2021 Jun;9(11):e14835. doi: 10.14814/phy2.14835. Physiol Rep. 2021. PMID: 34111326 Free PMC article.
References
-
- Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J Biol Chem 275: 28810–28815, 2000. - PubMed
-
- Beltran-Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post-translationally modified by arginine methylation. J Proteome Res 10: 3712–3719, 2011. - PubMed
-
- Beltran-Alvarez P, Tarradas A, Chiva C, Pérez-Serra A, Batlle M, Pérez-Villa F, Schulte U, Sabidó E, Brugada R, Pagans S. Identification of N-terminal protein acetylation and arginine methylation of the voltage-gated sodium channel in end-stage heart failure human heart. J Mol Cell Cardiol 76: 126–129, 2014. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous