

DNP, mitochondrial uncoupling, and neuroprotection: A little dab'll do ya
- PMID: 27599210
- PMCID: PMC5337177
- DOI: 10.1016/j.jalz.2016.08.001
DNP, mitochondrial uncoupling, and neuroprotection: A little dab'll do ya
Abstract
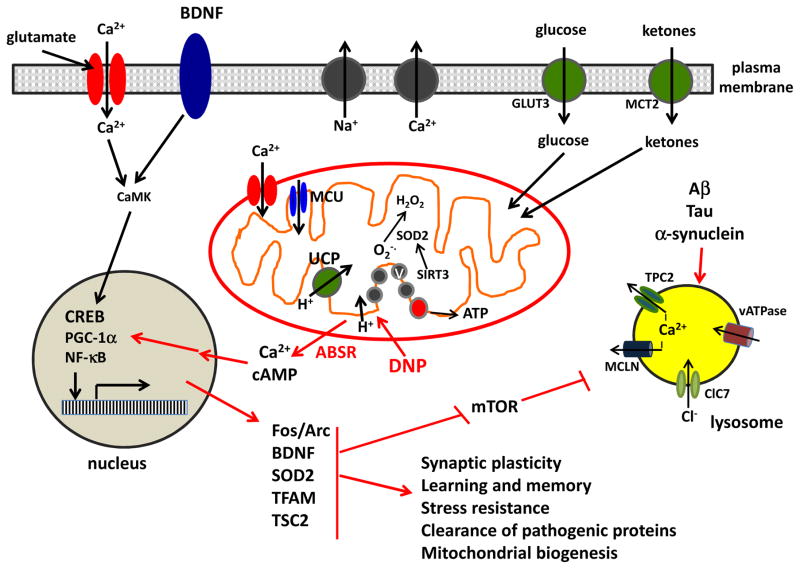
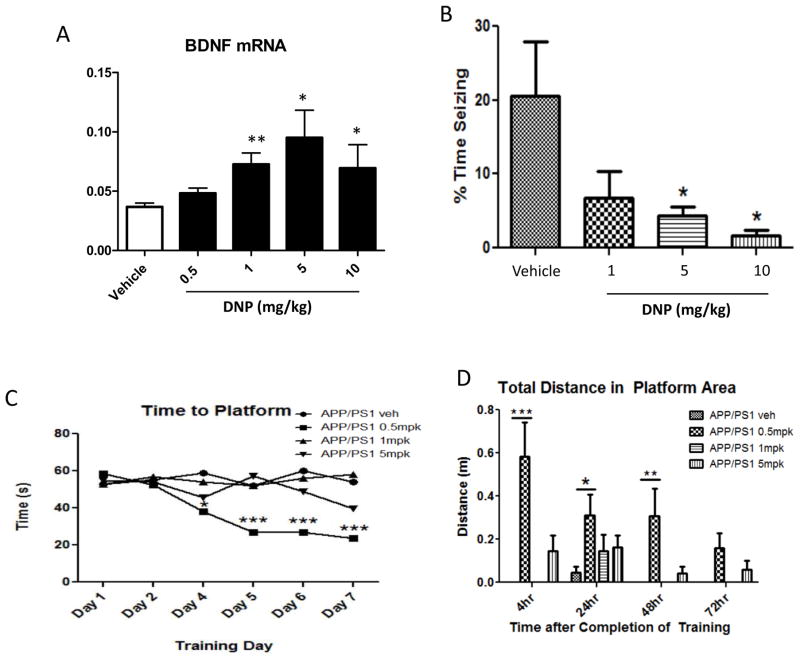
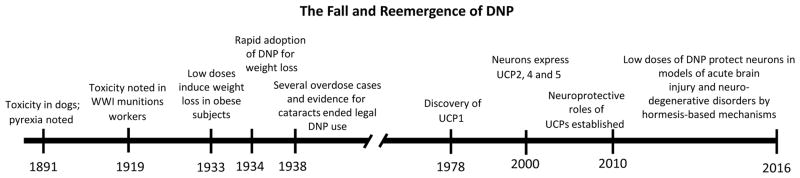
Recent findings have elucidated roles for mitochondrial uncoupling proteins (UCPs) in neuronal plasticity and resistance to metabolic and oxidative stress. UCPs are induced by bioenergetic challenges such as caloric restriction and exercise and may protect neurons against dysfunction and degeneration. The pharmacological uncoupler 2,4-dinitrophenol (DNP), which was once prescribed to >100,000 people as a treatment for obesity, stimulates several adaptive cellular stress-response signaling pathways in neurons including those involving the brain-derived neurotrophic factor (BDNF), the transcription factor cyclic AMP response element-binding protein (CREB), and autophagy. Preclinical data show that low doses of DNP can protect neurons and improve functional outcome in animal models of Alzheimer's and Parkinson's diseases, epilepsy, and cerebral ischemic stroke. Repurposing of DNP and the development of novel uncoupling agents with hormetic mechanisms of action provide opportunities for new breakthrough therapeutic interventions in a range of acute and chronic insidious neurodegenerative/neuromuscular conditions, all paradoxically at body weight-preserving doses.
Keywords: BDNF; CREB; DNP; Hormesis; Mitochondrial uncoupling; Parkinson's disease; Synaptic plasticity.
Published by Elsevier Inc.
Figures

Similar articles
-
The mitochondrial uncoupler DNP triggers brain cell mTOR signaling network reprogramming and CREB pathway up-regulation.J Neurochem. 2015 Aug;134(4):677-92. doi: 10.1111/jnc.13176. Epub 2015 Jun 19. J Neurochem. 2015. PMID: 26010875 Free PMC article.
-
2,4 Dinitrophenol as Medicine.Cells. 2019 Mar 23;8(3):280. doi: 10.3390/cells8030280. Cells. 2019. PMID: 30909602 Free PMC article. Review.
-
Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance.Free Radic Biol Med. 2017 Jan;102:203-216. doi: 10.1016/j.freeradbiomed.2016.11.045. Epub 2016 Nov 29. Free Radic Biol Med. 2017. PMID: 27908782 Free PMC article. Review.
-
Neuroprotective effects of 2,4-dinitrophenol in an acute model of Parkinson's disease.Brain Res. 2017 May 15;1663:184-193. doi: 10.1016/j.brainres.2017.03.018. Epub 2017 Mar 18. Brain Res. 2017. PMID: 28322751
-
The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia.J Neurochem. 2005 Sep;94(6):1676-84. doi: 10.1111/j.1471-4159.2005.03328.x. Epub 2005 Jul 25. J Neurochem. 2005. PMID: 16045446
Cited by
-
Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer's Disease.Int J Mol Sci. 2021 May 3;22(9):4850. doi: 10.3390/ijms22094850. Int J Mol Sci. 2021. PMID: 34063708 Free PMC article. Review.
-
6-Amino[1,2,5]oxadiazolo[3,4-b]pyrazin-5-ol Derivatives as Efficacious Mitochondrial Uncouplers in STAM Mouse Model of Nonalcoholic Steatohepatitis.J Med Chem. 2020 Jun 11;63(11):6203-6224. doi: 10.1021/acs.jmedchem.0c00542. Epub 2020 May 27. J Med Chem. 2020. PMID: 32392051 Free PMC article.
-
Mild pentachlorophenol-mediated uncoupling of mitochondria depletes ATP but does not cause an oxidized redox state or dopaminergic neurodegeneration in Caenorhabditis elegans.Curr Res Toxicol. 2022 Aug 2;3:100084. doi: 10.1016/j.crtox.2022.100084. eCollection 2022. Curr Res Toxicol. 2022. PMID: 35957653 Free PMC article.
-
2,4 DNP improves motor function, preserves medium spiny neuronal identity, and reduces oxidative stress in a mouse model of Huntington's disease.Exp Neurol. 2017 Jul;293:83-90. doi: 10.1016/j.expneurol.2017.03.020. Epub 2017 Mar 28. Exp Neurol. 2017. PMID: 28359739 Free PMC article.
-
Prodrug Therapies for Infectious and Neurodegenerative Diseases.Pharmaceutics. 2022 Feb 26;14(3):518. doi: 10.3390/pharmaceutics14030518. Pharmaceutics. 2022. PMID: 35335894 Free PMC article. Review.
References
-
- Barnard ND, Bush AI, Ceccarelli A, Cooper J, de Jager CA, Erickson KI, Fraser G, Kesler S, Levin SM, Lucey B, Morris MC, Squitti R. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol Aging. 2014;35:S74–78. - PubMed
-
- Yaffe K, Hoang TD, Byers AL, Barnes DE, Friedl KE. Lifestyle and health-related risk factors and risk of cognitive aging among older veterans. Alzheimers Dement. 2014;10:S111–121. - PubMed
-
- Duan W, Mattson MP. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J Neurosci Res. 1999;57:195–206. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
