

The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress
- PMID: 27556400
- PMCID: PMC4996521
- DOI: 10.1371/journal.ppat.1005825
The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress
Abstract
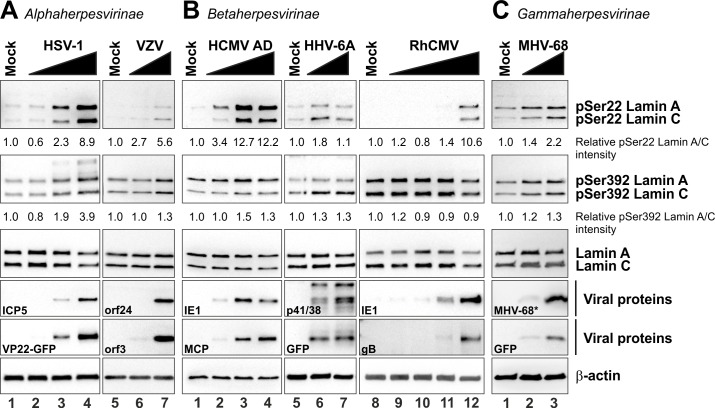
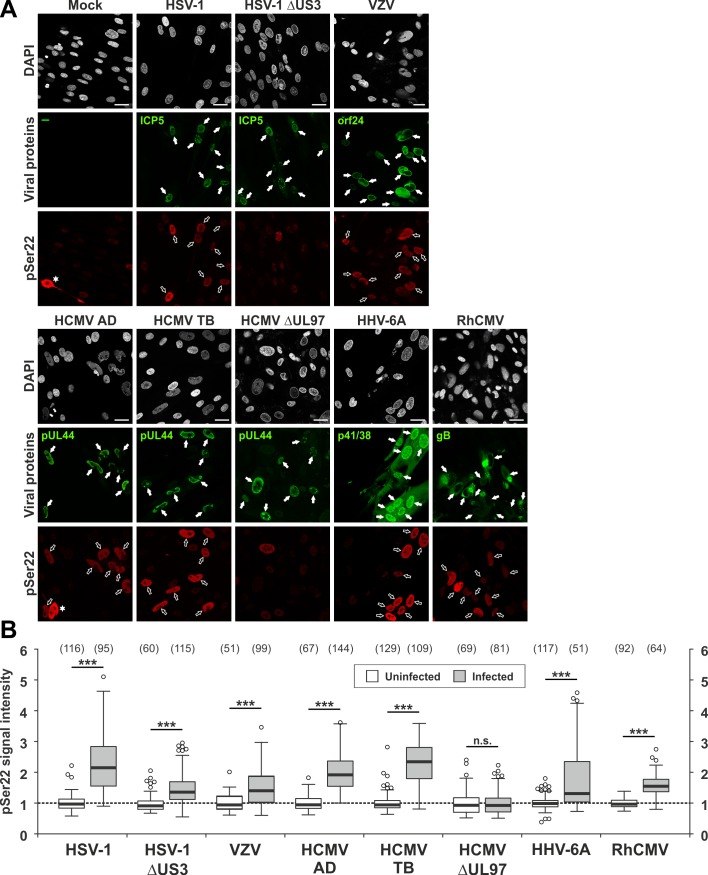
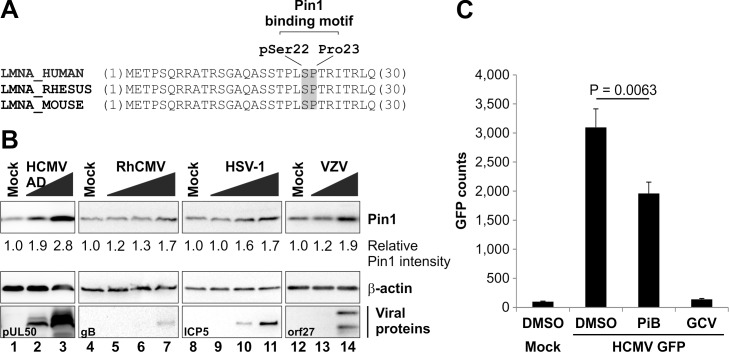
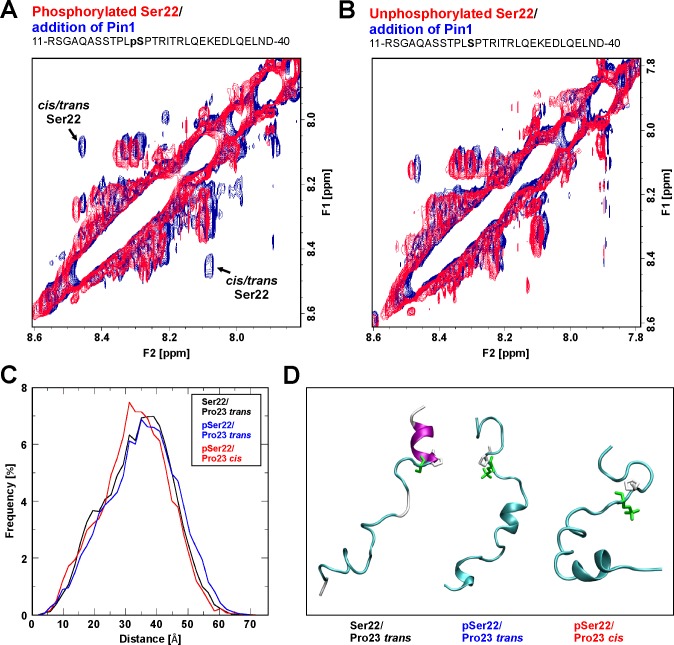
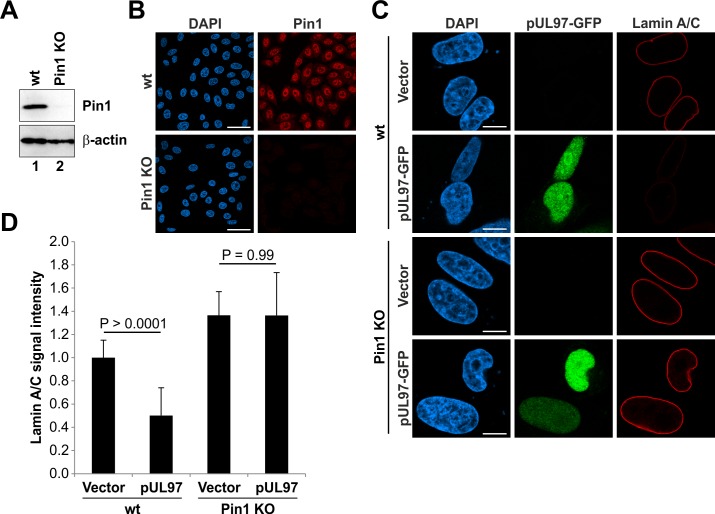
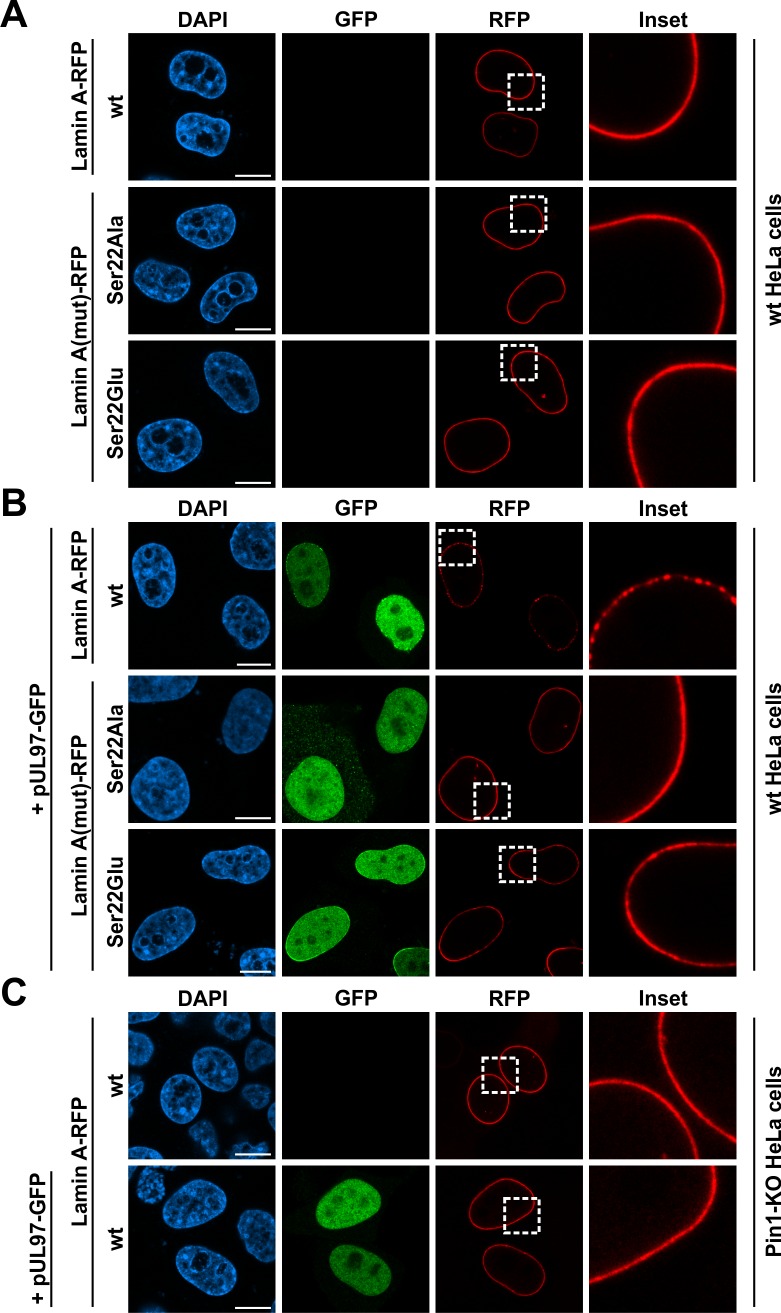
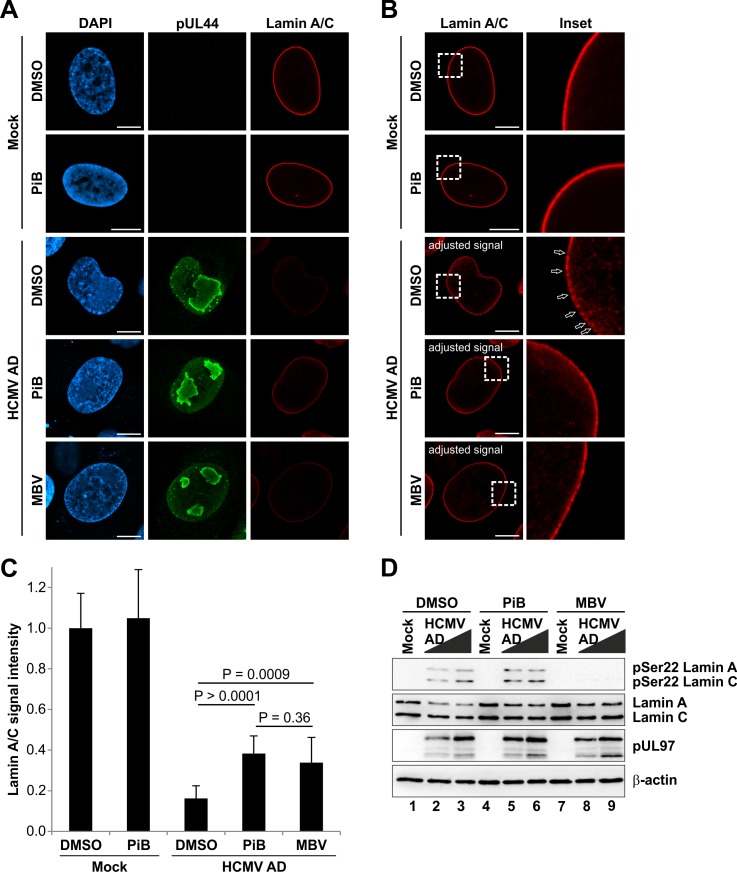
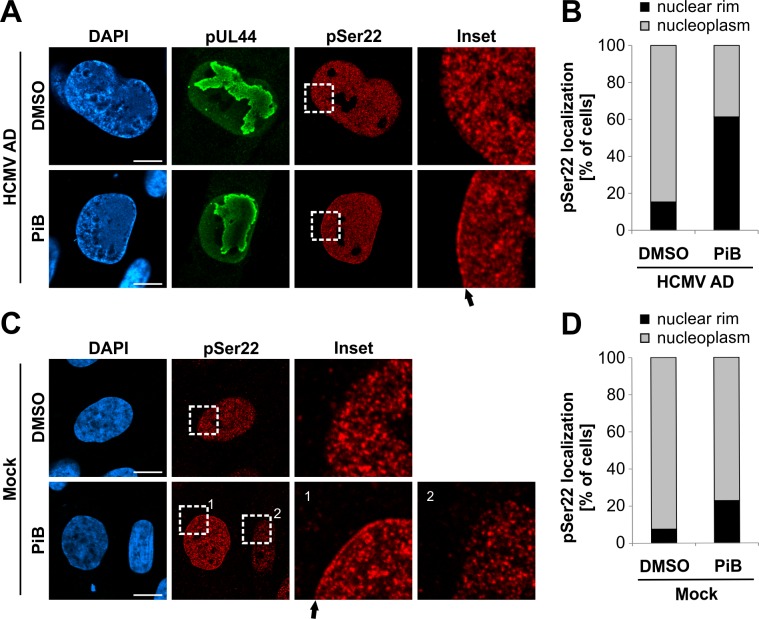
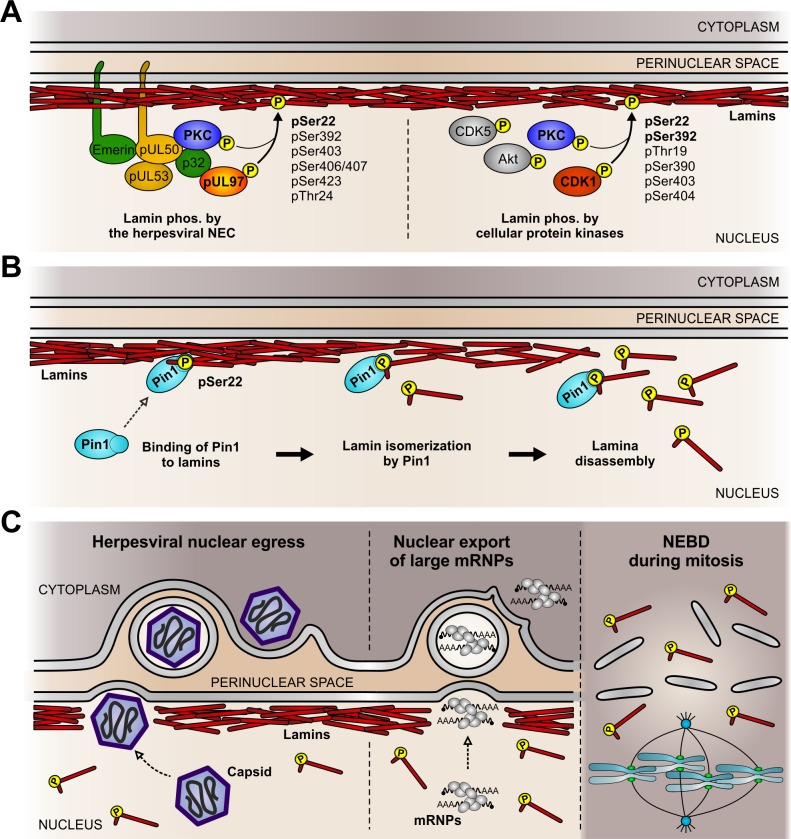
The nuclear lamina lines the inner nuclear membrane providing a structural framework for the nucleus. Cellular processes, such as nuclear envelope breakdown during mitosis or nuclear export of large ribonucleoprotein complexes, are functionally linked to the disassembly of the nuclear lamina. In general, lamina disassembly is mediated by phosphorylation, but the precise molecular mechanism is still not completely understood. Recently, we suggested a novel mechanism for lamina disassembly during the nuclear egress of herpesviral capsids which involves the cellular isomerase Pin1. In this study, we focused on mechanistic details of herpesviral nuclear replication to demonstrate the general importance of Pin1 for lamina disassembly. In particular, Ser22-specific lamin phosphorylation consistently generates a Pin1-binding motif in cells infected with human and animal alpha-, beta-, and gammaherpesviruses. Using nuclear magnetic resonance spectroscopy, we showed that binding of Pin1 to a synthetic lamin peptide induces its cis/trans isomerization in vitro. A detailed bioinformatic evaluation strongly suggests that this structural conversion induces large-scale secondary structural changes in the lamin N-terminus. Thus, we concluded that a Pin1-induced conformational change of lamins may represent the molecular trigger responsible for lamina disassembly. Consistent with this concept, pharmacological inhibition of Pin1 activity blocked lamina disassembly in herpesvirus-infected fibroblasts and consequently impaired virus replication. In addition, a phospho-mimetic Ser22Glu lamin mutant was still able to form a regular lamina structure and overexpression of a Ser22-phosphorylating kinase did not induce lamina disassembly in Pin1 knockout cells. Intriguingly, this was observed in absence of herpesvirus infection proposing a broader importance of Pin1 for lamina constitution. Thus, our results suggest a functional model of similar events leading to disassembly of the nuclear lamina in response to herpesviral or inherent cellular stimuli. In essence, Pin1 represents a regulatory effector of lamina disassembly that promotes the nuclear pore-independent egress of herpesviral capsids.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus.J Biol Chem. 2010 Apr 30;285(18):13979-89. doi: 10.1074/jbc.M109.063628. Epub 2010 Mar 4. J Biol Chem. 2010. PMID: 20202933 Free PMC article.
-
The peptidyl-prolyl cis/trans isomerase Pin1 interacts with three early regulatory proteins of human cytomegalovirus.Virus Res. 2020 Aug;285:198023. doi: 10.1016/j.virusres.2020.198023. Epub 2020 May 16. Virus Res. 2020. PMID: 32428517
-
Regulatory roles of protein kinases in cytomegalovirus replication.Adv Virus Res. 2011;80:69-101. doi: 10.1016/B978-0-12-385987-7.00004-X. Adv Virus Res. 2011. PMID: 21762822 Review.
-
Extragenic Suppression of a Mutation in Herpes Simplex Virus 1 UL34 That Affects Lamina Disruption and Nuclear Egress.J Virol. 2016 Nov 14;90(23):10738-10751. doi: 10.1128/JVI.01544-16. Print 2016 Dec 1. J Virol. 2016. PMID: 27654296 Free PMC article.
-
Nuclear Egress of Herpesviruses: The Prototypic Vesicular Nucleocytoplasmic Transport.Adv Virus Res. 2016;94:81-140. doi: 10.1016/bs.aivir.2015.10.002. Epub 2016 Jan 29. Adv Virus Res. 2016. PMID: 26997591 Review.
Cited by
-
Cross-regulation of viral kinases with cyclin A secures shutoff of host DNA synthesis.Nat Commun. 2020 Sep 24;11(1):4845. doi: 10.1038/s41467-020-18542-1. Nat Commun. 2020. PMID: 32973148 Free PMC article.
-
'Getting Better'-Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism?Int J Mol Sci. 2024 Feb 29;25(5):2823. doi: 10.3390/ijms25052823. Int J Mol Sci. 2024. PMID: 38474070 Free PMC article. Review.
-
Properties of Oligomeric Interaction of the Cytomegalovirus Core Nuclear Egress Complex (NEC) and Its Sensitivity to an NEC Inhibitory Small Molecule.Viruses. 2021 Mar 11;13(3):462. doi: 10.3390/v13030462. Viruses. 2021. PMID: 33799898 Free PMC article.
-
Roles of peptidyl prolyl isomerase Pin1 in viral propagation.Front Cell Dev Biol. 2022 Oct 25;10:1005325. doi: 10.3389/fcell.2022.1005325. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36393854 Free PMC article. Review.
-
Assessment of Covalently Binding Warhead Compounds in the Validation of the Cytomegalovirus Nuclear Egress Complex as an Antiviral Target.Cells. 2023 Apr 14;12(8):1162. doi: 10.3390/cells12081162. Cells. 2023. PMID: 37190072 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
