

TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells
- PMID: 27503209
- PMCID: PMC5070654
- DOI: 10.4049/jimmunol.1502702
TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells
Abstract
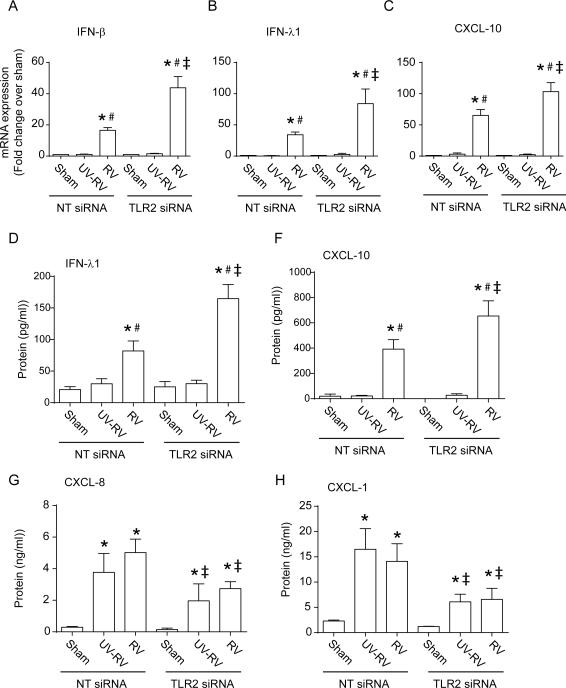
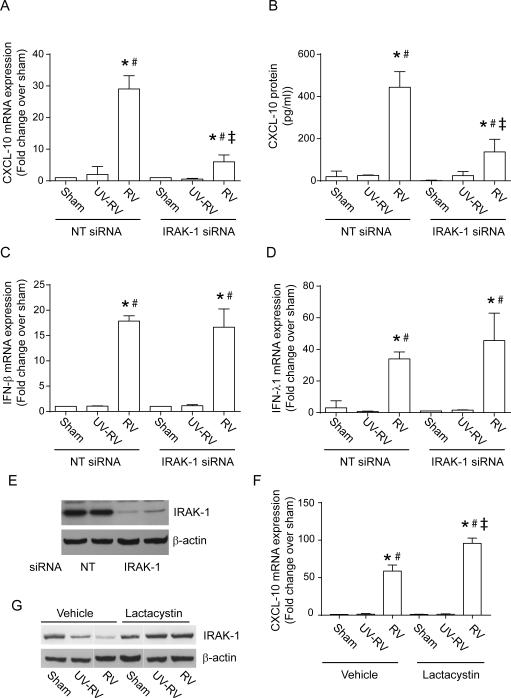
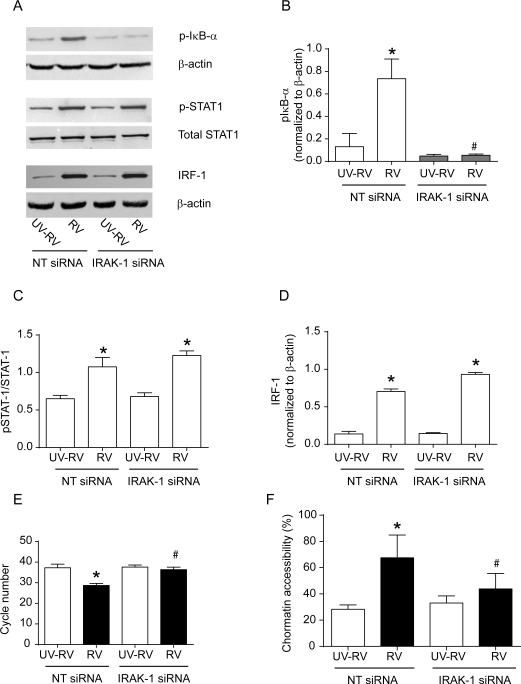
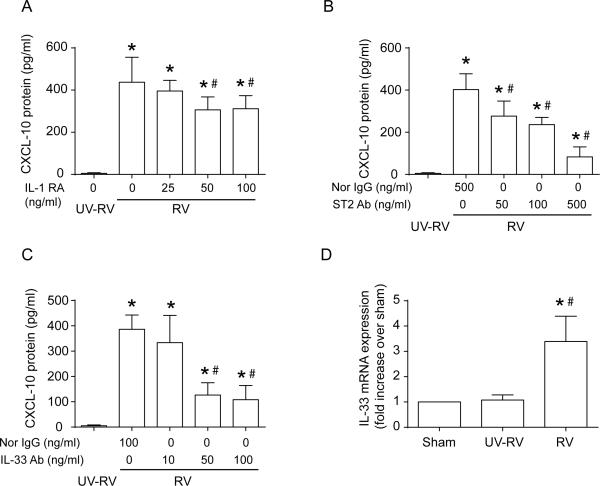
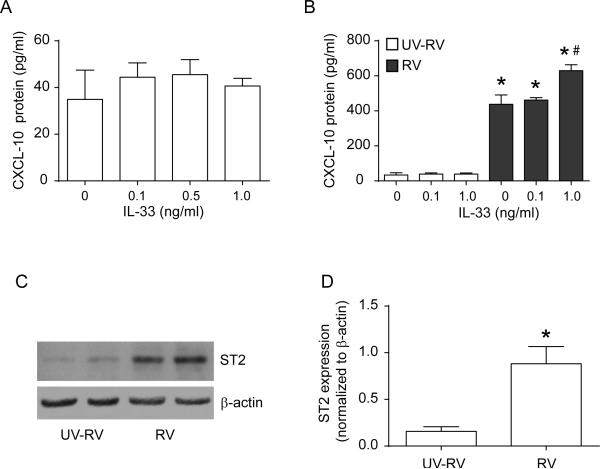
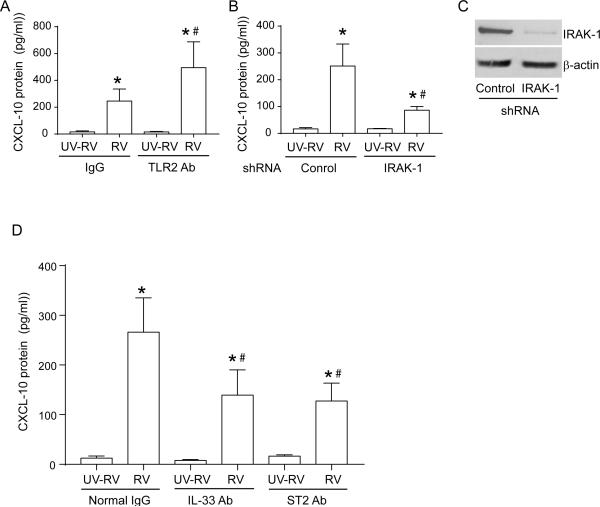
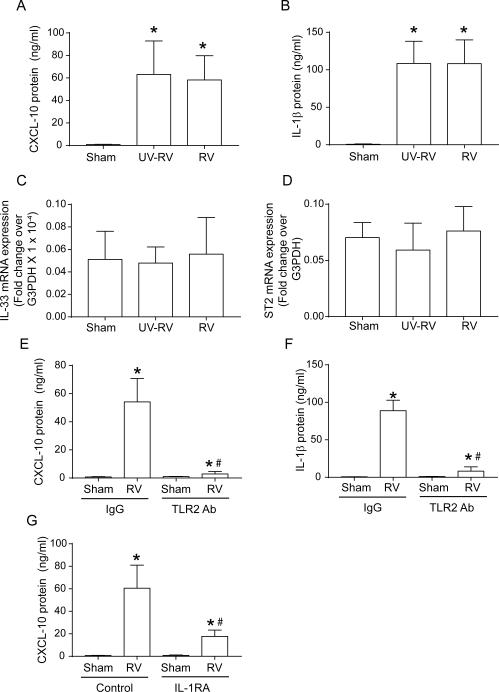
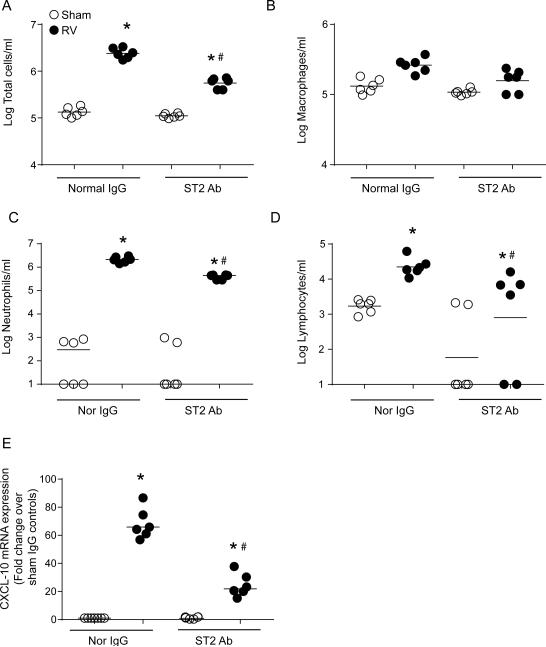
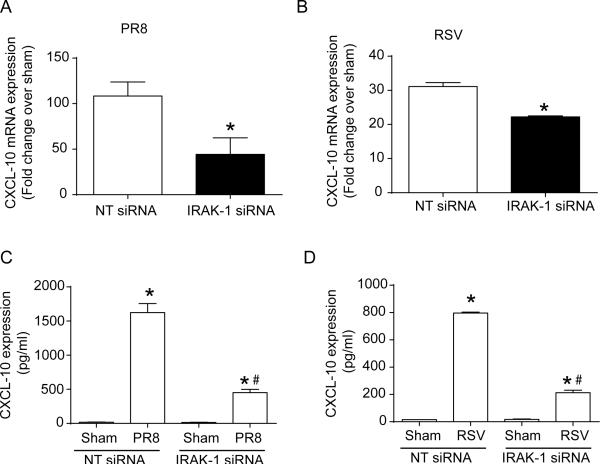
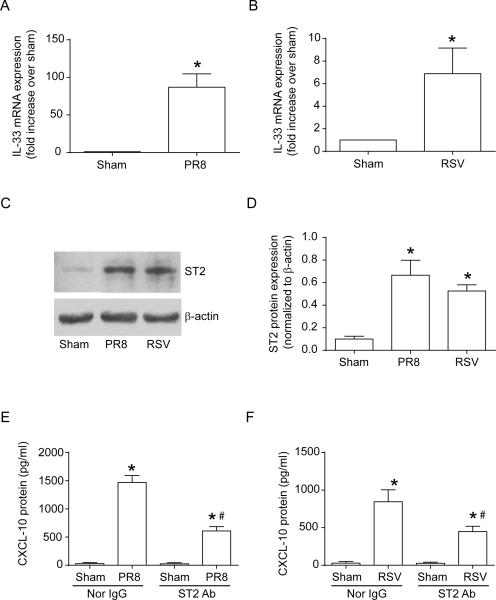
Airway epithelial cells are the major target for rhinovirus (RV) infection and express proinflammatory chemokines and antiviral cytokines that play a role in innate immunity. Previously, we demonstrated that RV interaction with TLR2 causes ILR-associated kinase-1 (IRAK-1) depletion in both airway epithelial cells and macrophages. Further, IRAK-1 degradation caused by TLR2 activation was shown to inhibit ssRNA-induced IFN expression in dendritic cells. Therefore, in this study, we examined the role of TLR2 and IRAK-1 in RV-induced IFN-β, IFN-λ1, and CXCL-10, which require signaling by viral RNA. In airway epithelial cells, blocking TLR2 enhanced RV-induced expression of IFNs and CXCL-10. By contrast, IRAK-1 inhibition abrogated RV-induced expression of CXCL-10, but not IFNs in these cells. Neutralization of IL-33 or its receptor, ST2, which requires IRAK-1 for signaling, inhibited RV-stimulated CXCL-10 expression. In addition, RV induced expression of both ST2 and IL-33 in airway epithelial cells. In macrophages, however, RV-stimulated CXCL-10 expression was primarily dependent on TLR2/IL-1R. Interestingly, in a mouse model of RV infection, blocking ST2 not only attenuated RV-induced CXCL-10, but also lung inflammation. Finally, influenza- and respiratory syncytial virus-induced CXCL-10 was also found to be partially dependent on IL-33/ST2/IRAK-1 signaling in airway epithelial cells. Together, our results indicate that RV stimulates CXCL-10 expression via the IL-33/ST2 signaling axis, and that TLR2 signaling limits RV-induced CXCL-10 via IRAK-1 depletion at least in airway epithelial cells. To our knowledge, this is the first report to demonstrate the role of respiratory virus-induced IL-33 in the induction of CXCL-10 in airway epithelial cells.
Copyright © 2016 by The American Association of Immunologists, Inc.
Figures
Similar articles
-
Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma.Viruses. 2020 Nov 19;12(11):1328. doi: 10.3390/v12111328. Viruses. 2020. PMID: 33227953 Free PMC article. Review.
-
Rhinovirus attenuates non-typeable Hemophilus influenzae-stimulated IL-8 responses via TLR2-dependent degradation of IRAK-1.PLoS Pathog. 2012;8(10):e1002969. doi: 10.1371/journal.ppat.1002969. Epub 2012 Oct 4. PLoS Pathog. 2012. PMID: 23055935 Free PMC article.
-
Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis.Clin Sci (Lond). 2019 Apr 29;133(8):983-996. doi: 10.1042/CS20181088. Print 2019 Apr 30. Clin Sci (Lond). 2019. PMID: 30952808 Free PMC article.
-
Tollip Inhibits ST2 Signaling in Airway Epithelial Cells Exposed to Type 2 Cytokines and Rhinovirus.J Innate Immun. 2020;12(1):103-115. doi: 10.1159/000497072. Epub 2019 Mar 29. J Innate Immun. 2020. PMID: 30928973 Free PMC article.
-
Innate Immune Responses by Respiratory Viruses, Including Rhinovirus, During Asthma Exacerbation.Front Immunol. 2022 Jun 20;13:865973. doi: 10.3389/fimmu.2022.865973. eCollection 2022. Front Immunol. 2022. PMID: 35795686 Free PMC article. Review.
Cited by
-
Calcitriol attenuates TLR2/IL-33 signaling pathway to repress Th9 cell differentiation and potentially limits the pathophysiology of rheumatoid arthritis.Mol Cell Biochem. 2021 Jan;476(1):369-384. doi: 10.1007/s11010-020-03914-4. Epub 2020 Sep 23. Mol Cell Biochem. 2021. PMID: 32965596
-
Rhinovirus-Induced Modulation of Epithelial Phenotype: Role in Asthma.Viruses. 2020 Nov 19;12(11):1328. doi: 10.3390/v12111328. Viruses. 2020. PMID: 33227953 Free PMC article. Review.
-
Rhinovirus and Innate Immune Function of Airway Epithelium.Front Cell Infect Microbiol. 2020 Jun 19;10:277. doi: 10.3389/fcimb.2020.00277. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 32637363 Free PMC article. Review.
-
IL-33: biological properties, functions, and roles in airway disease.Immunol Rev. 2017 Jul;278(1):173-184. doi: 10.1111/imr.12552. Immunol Rev. 2017. PMID: 28658560 Free PMC article. Review.
-
Priming With Rhinovirus Protects Mice Against a Lethal Pulmonary Coronavirus Infection.Front Immunol. 2022 May 30;13:886611. doi: 10.3389/fimmu.2022.886611. eCollection 2022. Front Immunol. 2022. PMID: 35711419 Free PMC article.
References
-
- Contoli M, Marku B, Conti V, Saturni S, Caramori G, Papi A. Viral infections in exacerbations of asthma and chronic obstructive pulmonary disease. Minerva Med. 2009;100:467–478. - PubMed
-
- Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, Curtis JL, Martinez FJ, Hershenson MB, Sajjan U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:332–340. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources