

More to Life than NF-κB in TNFR1 Signaling
- PMID: 27424290
- PMCID: PMC5076853
- DOI: 10.1016/j.it.2016.06.002
More to Life than NF-κB in TNFR1 Signaling
Abstract
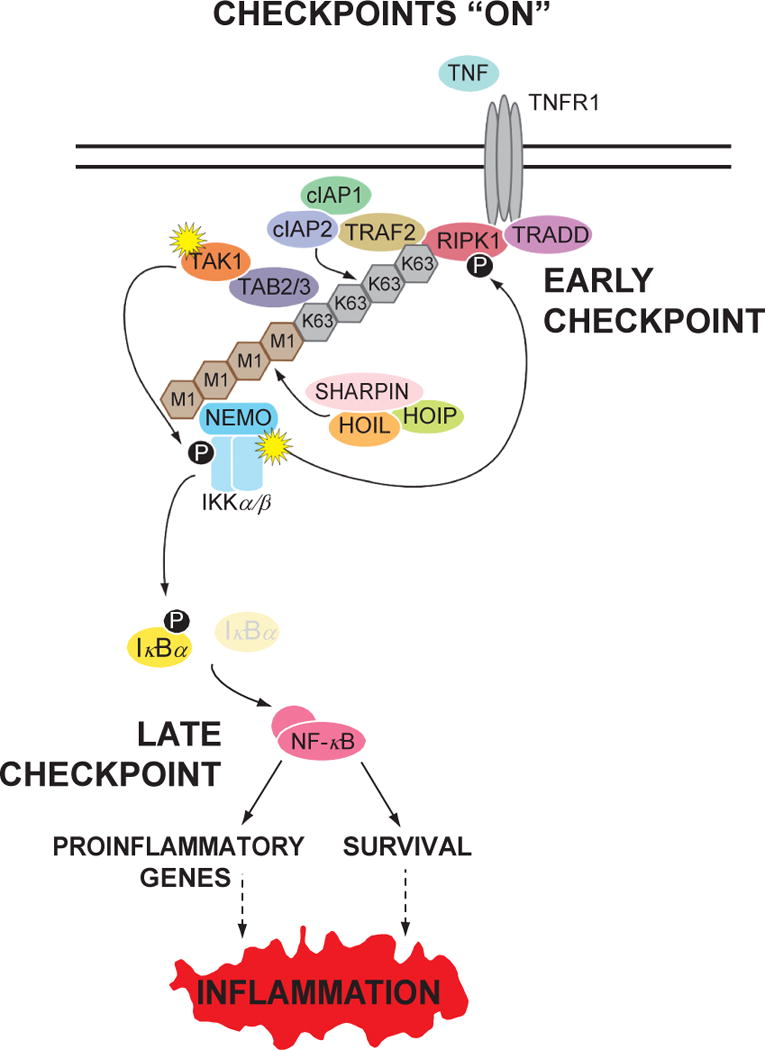
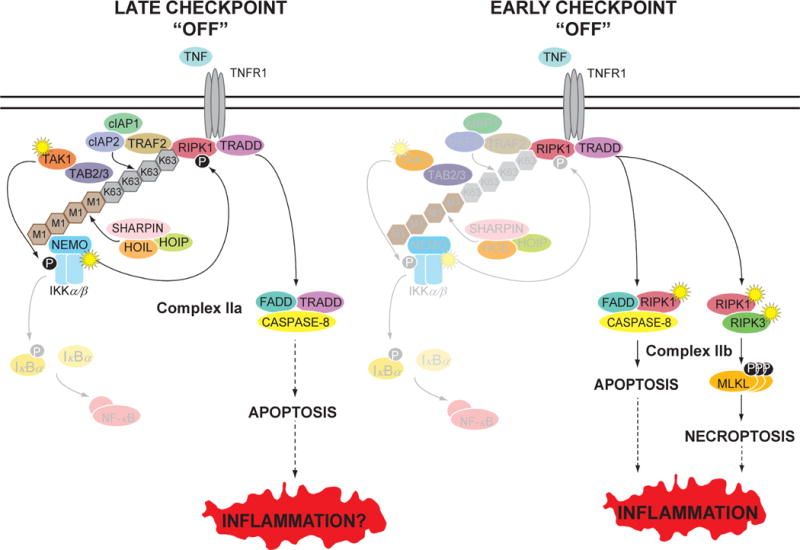
TNF is a master proinflammatory cytokine whose pathogenic role in inflammatory disorders has long been attributed to induction of proinflammatory mediators. TNF also activates cell survival and death pathways, and recent studies demonstrated that TNF also causes inflammation by inducing cell death. The default response of most cells to TNF is survival and NF-κB-mediated upregulation of prosurvival molecules is a well-documented protective mechanism downstream of TNFR1. Recent studies revealed the existence of an NF-κB-independent cell death checkpoint that restricts cell demise by inactivating RIPK1. Disruption of this checkpoint leads to RIPK1 kinase-dependent death and causes inflammation in vivo. These revelations bring complexity to the control of TNF-induced cell death, and suggest clinical benefit of RIPK1 inhibitors in TNF-driven human inflammatory disorders.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling.Mol Cell. 2015 Oct 1;60(1):63-76. doi: 10.1016/j.molcel.2015.07.032. Epub 2015 Sep 3. Mol Cell. 2015. PMID: 26344099
-
StIKKing it to a death kinase: IKKs prevent TNF-α-induced cell death by phosphorylating RIPK1.Cytokine. 2016 Feb;78:47-50. doi: 10.1016/j.cyto.2015.10.014. Epub 2015 Nov 28. Cytokine. 2016. PMID: 26630177
-
TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death.Biochem Pharmacol. 2016 Sep 15;116:1-10. doi: 10.1016/j.bcp.2016.03.009. Epub 2016 Mar 16. Biochem Pharmacol. 2016. PMID: 26993379 Review.
-
The NF-kappaB transcription factor pathway as a therapeutic target in cancer: methods for detection of NF-kappaB activity.Methods Mol Biol. 2009;512:169-207. doi: 10.1007/978-1-60327-530-9_10. Methods Mol Biol. 2009. PMID: 19347278
-
TNFR1-activated NF-κB signal transduction: regulation by the ubiquitin/proteasome system.Curr Opin Chem Biol. 2014 Dec;23:71-7. doi: 10.1016/j.cbpa.2014.10.011. Curr Opin Chem Biol. 2014. PMID: 25461388 Review.
Cited by
-
Editorial: TNFR Superfamily Oligomerization and Signaling.Front Cell Dev Biol. 2021 Apr 20;9:682472. doi: 10.3389/fcell.2021.682472. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33959618 Free PMC article. No abstract available.
-
The mechanistic functional landscape of retinitis pigmentosa: a machine learning-driven approach to therapeutic target discovery.J Transl Med. 2024 Feb 6;22(1):139. doi: 10.1186/s12967-024-04911-7. J Transl Med. 2024. PMID: 38321543 Free PMC article.
-
OTULIN maintains skin homeostasis by controlling keratinocyte death and stem cell identity.Nat Commun. 2021 Oct 8;12(1):5913. doi: 10.1038/s41467-021-25944-2. Nat Commun. 2021. PMID: 34625556 Free PMC article.
-
Cell death in skin function, inflammation, and disease.Biochem J. 2022 Aug 12;479(15):1621-1651. doi: 10.1042/BCJ20210606. Biochem J. 2022. PMID: 35929827 Free PMC article. Review.
-
Immune dysregulation in SHARPIN-deficient mice is dependent on CYLD-mediated cell death.Proc Natl Acad Sci U S A. 2021 Dec 14;118(50):e2001602118. doi: 10.1073/pnas.2001602118. Proc Natl Acad Sci U S A. 2021. PMID: 34887354 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
