

Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD)
- PMID: 27389053
- PMCID: PMC5214979
- DOI: 10.1038/leu.2016.160
Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD)
Abstract
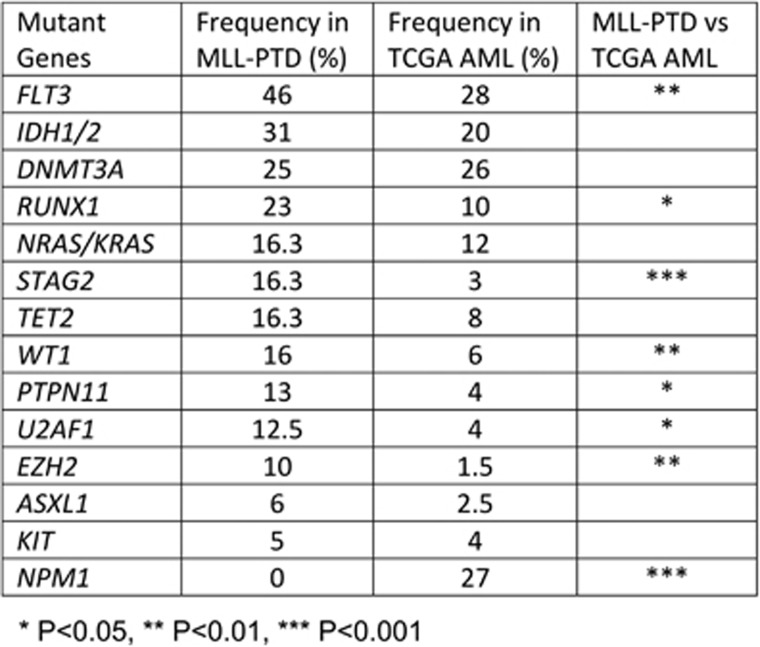
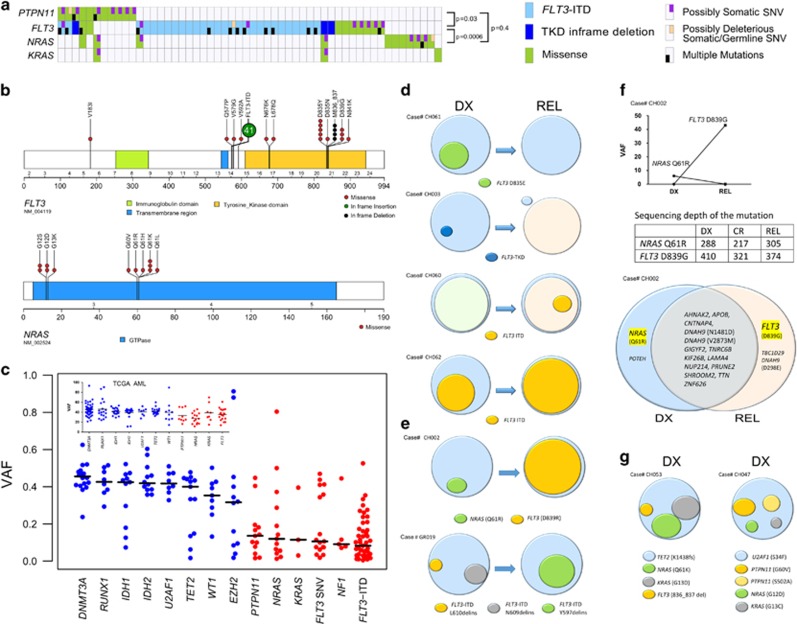
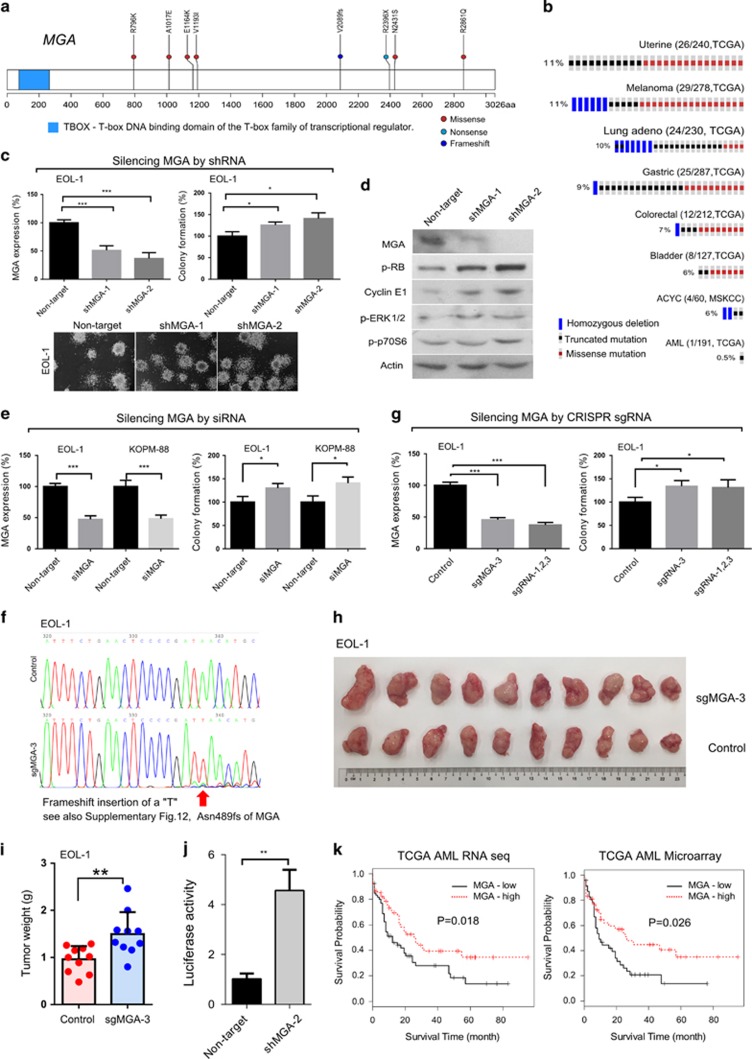
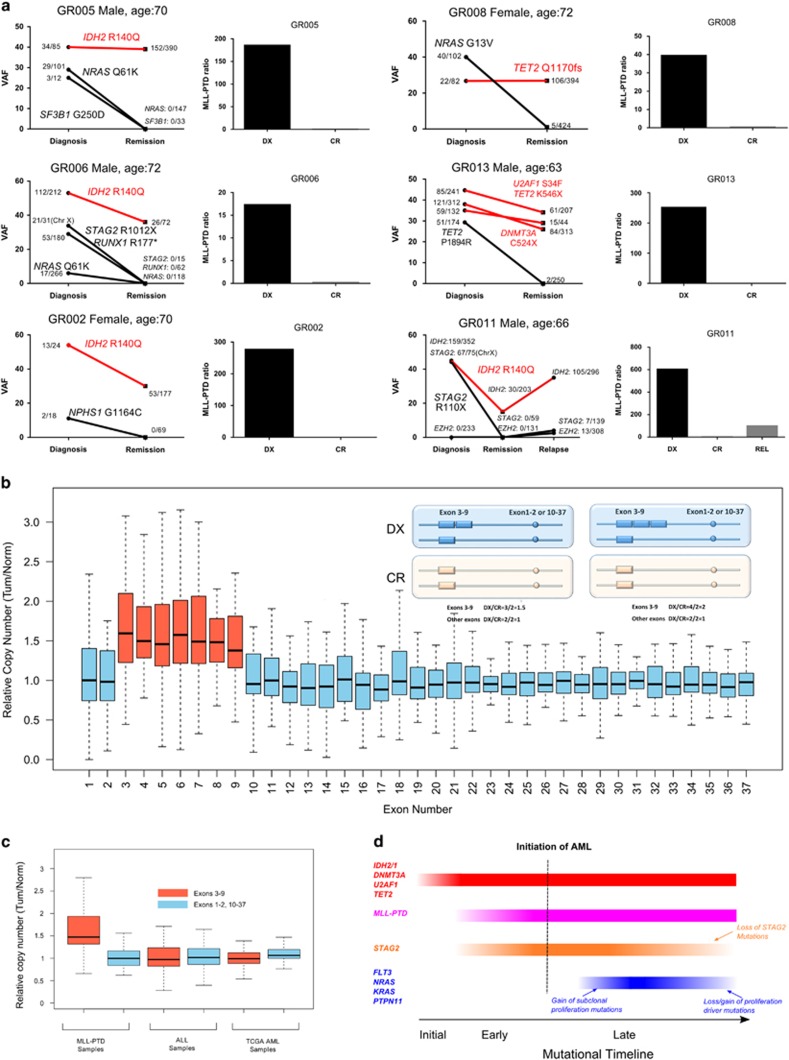
Partial tandem duplication of MLL (MLL-PTD) characterizes acute myeloid leukemia (AML) patients often with a poor prognosis. To understand the order of occurrence of MLL-PTD in relation to other major AML mutations and to identify novel mutations that may be present in this unique AML molecular subtype, exome and targeted sequencing was performed on 85 MLL-PTD AML samples using HiSeq-2000. Genes involved in the cohesin complex (STAG2), a splicing factor (U2AF1) and a poorly studied gene, MGA were recurrently mutated, whereas NPM1, one of the most frequently mutated AML gene, was not mutated in MLL-PTD patients. Interestingly, clonality analysis suggests that IDH2/1, DNMT3A, U2AF1 and TET2 mutations are clonal and occur early, and MLL-PTD likely arises after these initial mutations. Conversely, proliferative mutations (FLT3, RAS), typically appear later, are largely subclonal and tend to be unstable. This study provides important insights for understanding the relative importance of different mutations for defining a targeted therapeutic strategy for MLL-PTD AML patients.
Conflict of interest statement
Torsten Haferlach is employed by and partly owns the MLL Munich Leukemia Laboratory. Tamara Alpermann and Manja Meggendorfer are employed by the MLL Munich Leukemia Laboratory. The remaining authors declare no competing financial interests.
Figures


Similar articles
-
High frequency of additional gene mutations in acute myeloid leukemia with MLL partial tandem duplication: DNMT3A mutation is associated with poor prognosis.Oncotarget. 2015 Oct 20;6(32):33217-25. doi: 10.18632/oncotarget.5202. Oncotarget. 2015. PMID: 26375248 Free PMC article.
-
Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia.Genes Chromosomes Cancer. 2003 Jul;37(3):237-51. doi: 10.1002/gcc.10219. Genes Chromosomes Cancer. 2003. PMID: 12759922
-
Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias.Blood. 2012 Aug 2;120(5):1130-6. doi: 10.1182/blood-2012-03-415067. Epub 2012 Jun 6. Blood. 2012. PMID: 22674806 Free PMC article.
-
The MLL partial tandem duplication in acute myeloid leukaemia.Br J Haematol. 2006 Nov;135(4):438-49. doi: 10.1111/j.1365-2141.2006.06301.x. Epub 2006 Sep 11. Br J Haematol. 2006. PMID: 16965385 Review.
-
Prognostic implications of gene mutations in acute myeloid leukemia with normal cytogenetics.Semin Oncol. 2008 Aug;35(4):346-55. doi: 10.1053/j.seminoncol.2008.04.005. Semin Oncol. 2008. PMID: 18692685 Review.
Cited by
-
A comprehensive review of genetic alterations and molecular targeted therapies for the implementation of personalized medicine in acute myeloid leukemia.J Mol Med (Berl). 2020 Aug;98(8):1069-1091. doi: 10.1007/s00109-020-01944-5. Epub 2020 Jul 3. J Mol Med (Berl). 2020. PMID: 32620999 Review.
-
The Progress of Next Generation Sequencing in the Assessment of Myeloid Malignancies.Balkan Med J. 2019 Feb 28;36(2):78-87. doi: 10.4274/balkanmedj.galenos.2018.2018.1195. Epub 2018 Sep 25. Balkan Med J. 2019. PMID: 30251956 Free PMC article.
-
Gene rearrangements of MLL and RUNX1 sporadically occur in normal CD34+ cells under cytokine stimulation.Cancer Sci. 2020 May;111(5):1851-1855. doi: 10.1111/cas.14392. Epub 2020 Apr 14. Cancer Sci. 2020. PMID: 32216001 Free PMC article.
-
Clinical implications of molecular markers in acute myeloid leukemia.Eur J Haematol. 2019 Jan;102(1):20-35. doi: 10.1111/ejh.13172. Epub 2018 Oct 23. Eur J Haematol. 2019. PMID: 30203623 Free PMC article. Review.
-
Executable cancer models: successes and challenges.Nat Rev Cancer. 2020 Jun;20(6):343-354. doi: 10.1038/s41568-020-0258-x. Epub 2020 Apr 27. Nat Rev Cancer. 2020. PMID: 32341552 Review.
References
-
- Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006; 368: 1894–1907. - PubMed
-
- Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia 2014; 28: 1586–1595. - PubMed
-
- Dohner K, Tobis K, Ulrich R, Frohling S, Benner A, Schlenk RF et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol 2002; 20: 3254–3261. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
