

Clonal evolution of glioblastoma under therapy
- PMID: 27270107
- PMCID: PMC5627776
- DOI: 10.1038/ng.3590
Clonal evolution of glioblastoma under therapy
Abstract
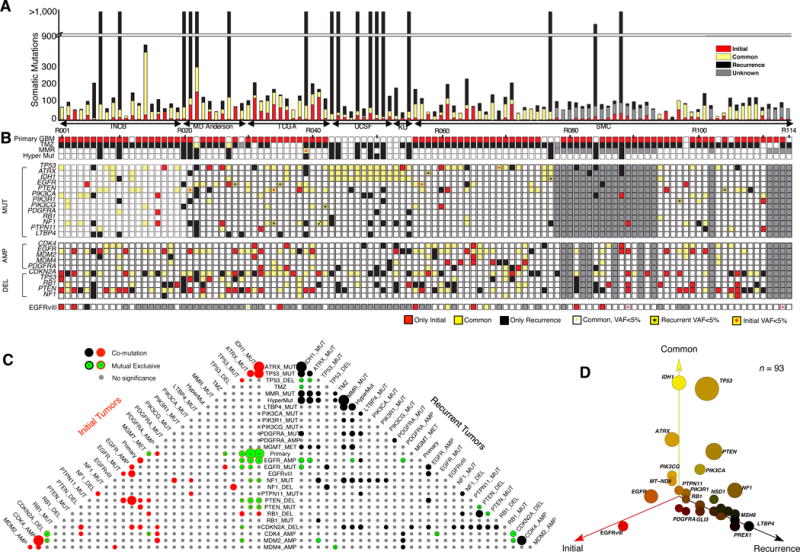
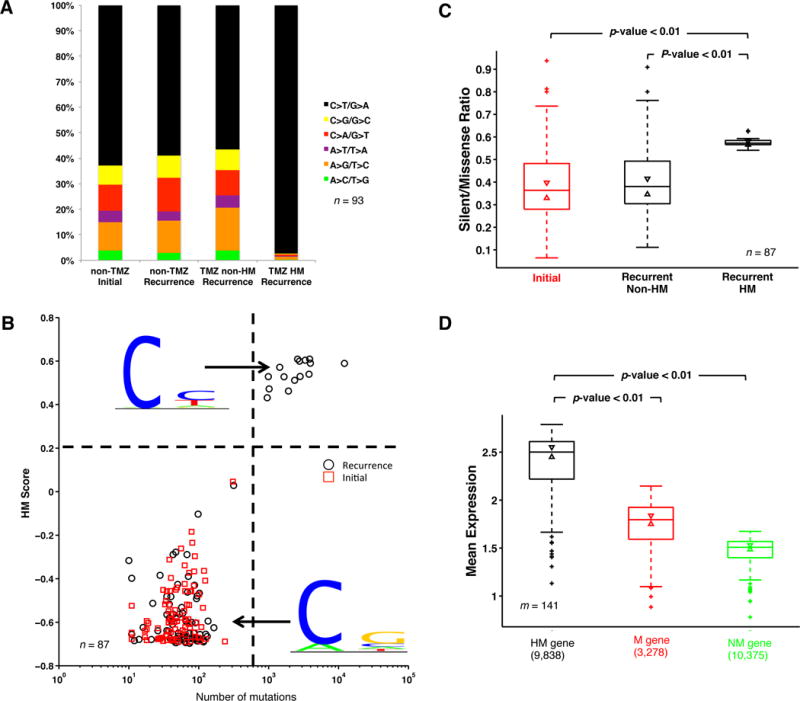
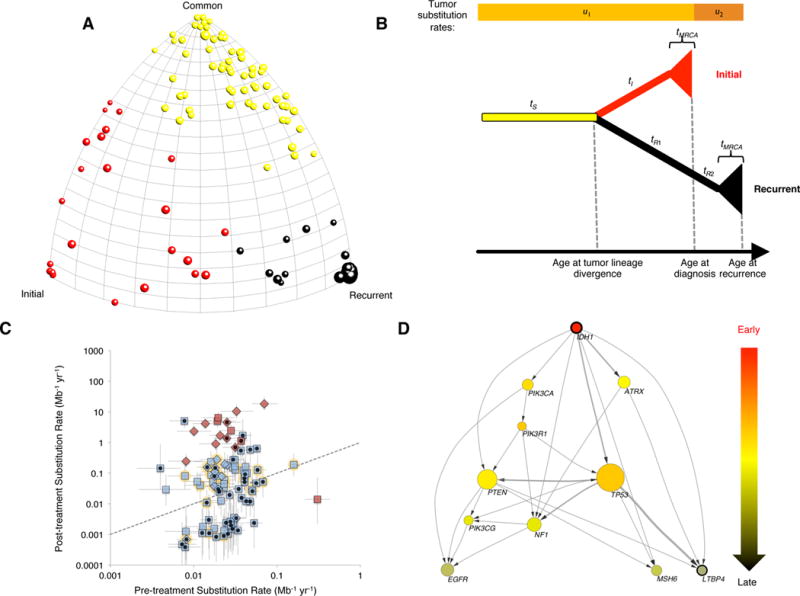
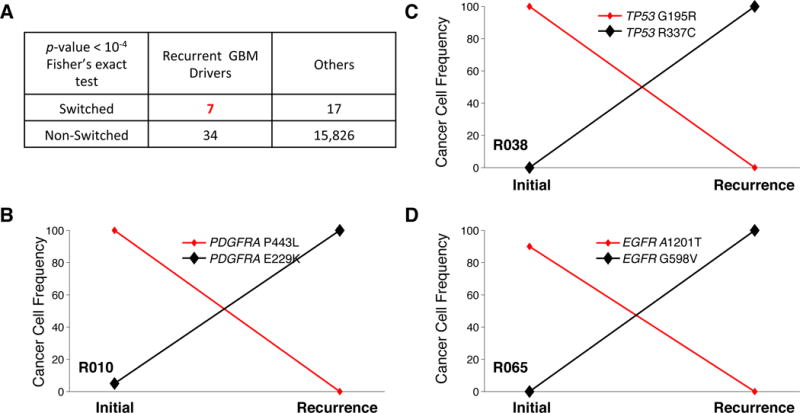
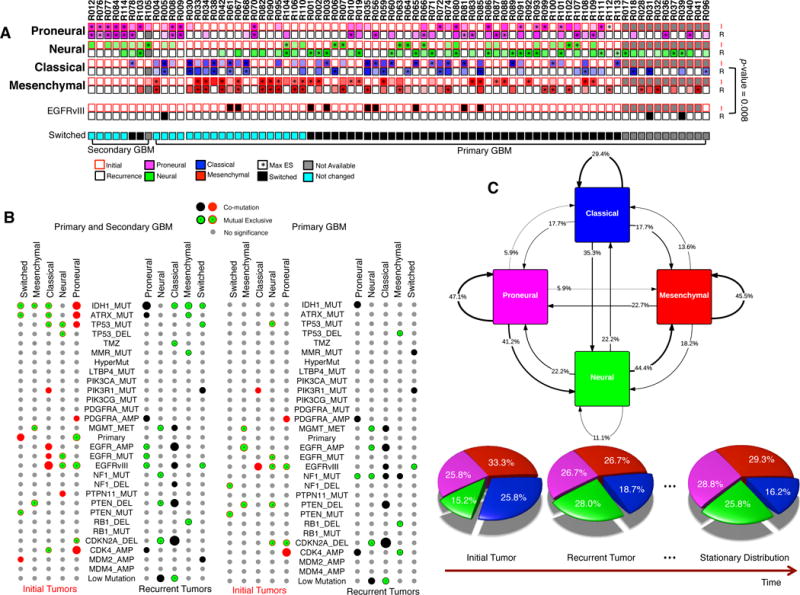
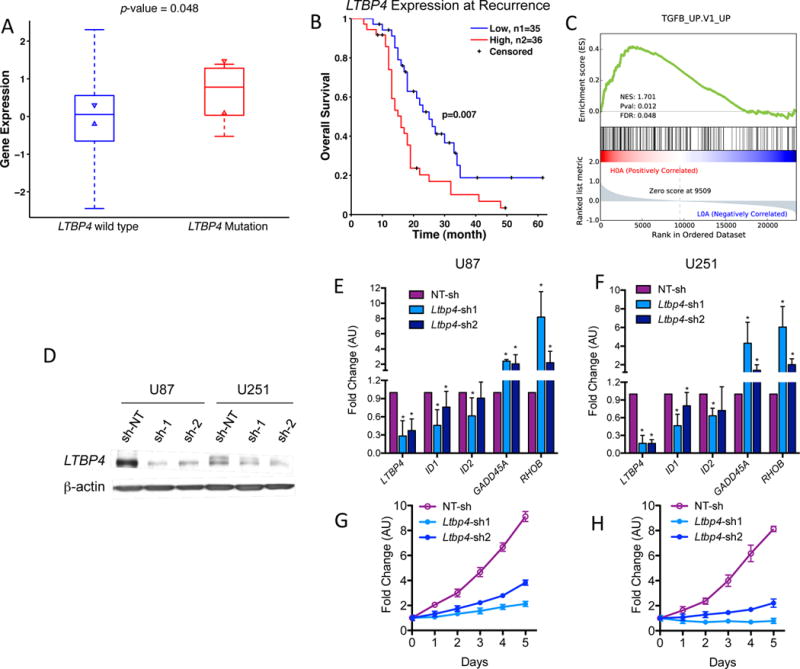
Glioblastoma (GBM) is the most common and aggressive primary brain tumor. To better understand how GBM evolves, we analyzed longitudinal genomic and transcriptomic data from 114 patients. The analysis shows a highly branched evolutionary pattern in which 63% of patients experience expression-based subtype changes. The branching pattern, together with estimates of evolutionary rate, suggests that relapse-associated clones typically existed years before diagnosis. Fifteen percent of tumors present hypermutation at relapse in highly expressed genes, with a clear mutational signature. We find that 11% of recurrence tumors harbor mutations in LTBP4, which encodes a protein binding to TGF-β. Silencing LTBP4 in GBM cells leads to suppression of TGF-β activity and decreased cell proliferation. In recurrent GBM with wild-type IDH1, high LTBP4 expression is associated with worse prognosis, highlighting the TGF-β pathway as a potential therapeutic target in GBM.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy.Neuro Oncol. 2017 Mar 1;19(3):394-404. doi: 10.1093/neuonc/now189. Neuro Oncol. 2017. PMID: 27571882 Free PMC article.
-
Up-regulation of miR-370-3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression.Sci Rep. 2016 Sep 6;6:32972. doi: 10.1038/srep32972. Sci Rep. 2016. PMID: 27595933 Free PMC article.
-
MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma.Pediatr Blood Cancer. 2007 Apr;48(4):403-7. doi: 10.1002/pbc.20803. Pediatr Blood Cancer. 2007. PMID: 16609952
-
Treatment considerations for MGMT-unmethylated glioblastoma.Curr Neurol Neurosci Rep. 2015 Jan;15(1):507. doi: 10.1007/s11910-014-0507-z. Curr Neurol Neurosci Rep. 2015. PMID: 25394859 Review.
-
Treatment of elderly patients with glioblastoma: a systematic evidence-based analysis.JAMA Neurol. 2015 May;72(5):589-96. doi: 10.1001/jamaneurol.2014.3739. JAMA Neurol. 2015. PMID: 25822375 Review.
Cited by
-
Pan-cancer analysis of heterogeneity of tumor mutational burden and genomic mutation under treatment pressure.ESMO Open. 2024 Jul;9(7):103494. doi: 10.1016/j.esmoop.2024.103494. Epub 2024 Jul 8. ESMO Open. 2024. PMID: 38981309 Free PMC article.
-
Challenging Hurdles of Current Targeting in Glioblastoma: A Focus on Immunotherapeutic Strategies.Int J Mol Sci. 2021 Mar 28;22(7):3493. doi: 10.3390/ijms22073493. Int J Mol Sci. 2021. PMID: 33800593 Free PMC article. Review.
-
The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma.Cells. 2021 Feb 24;10(3):484. doi: 10.3390/cells10030484. Cells. 2021. PMID: 33668200 Free PMC article. Review.
-
On the Boundary of Exploratory Genomics and Translation in Sequential Glioblastoma.Int J Mol Sci. 2024 Jul 10;25(14):7564. doi: 10.3390/ijms25147564. Int J Mol Sci. 2024. PMID: 39062807 Free PMC article.
-
Long, Noncoding RNA Dysregulation in Glioblastoma.Cancers (Basel). 2021 Mar 31;13(7):1604. doi: 10.3390/cancers13071604. Cancers (Basel). 2021. PMID: 33807183 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
- T15 LM007079/LM/NLM NIH HHS/United States
- R01 CA185486/CA/NCI NIH HHS/United States
- R01 CA179044/CA/NCI NIH HHS/United States
- R01 CA131126/CA/NCI NIH HHS/United States
- R01 CA178546/CA/NCI NIH HHS/United States
- R01 GM117591/GM/NIGMS NIH HHS/United States
- TL1 TR000082/TR/NCATS NIH HHS/United States
- R01 CA101644/CA/NCI NIH HHS/United States
- T32 CA009503/CA/NCI NIH HHS/United States
- U54 CA193313/CA/NCI NIH HHS/United States
- UL1 TR000040/TR/NCATS NIH HHS/United States
- R01 NS061776/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
