

Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS) Motor Dysfunction Modeled in Mice
- PMID: 27255703
- PMCID: PMC5014696
- DOI: 10.1007/s12311-016-0797-6
Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS) Motor Dysfunction Modeled in Mice
Abstract
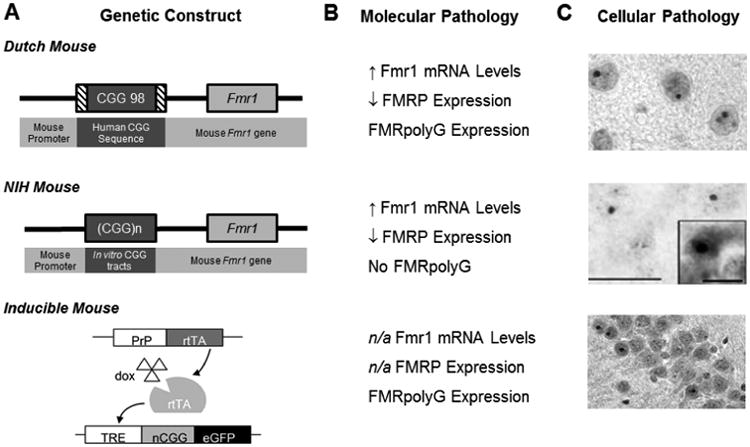
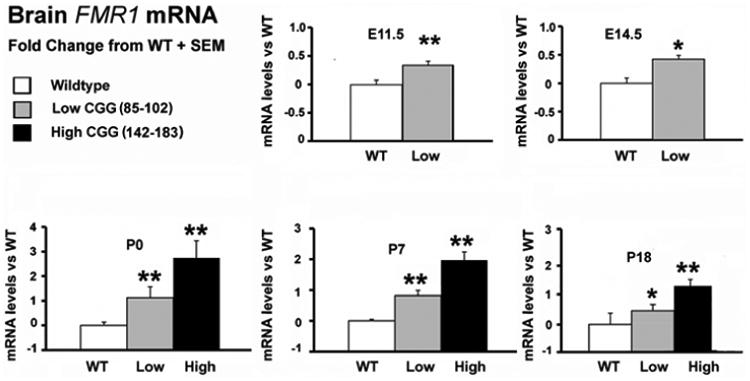
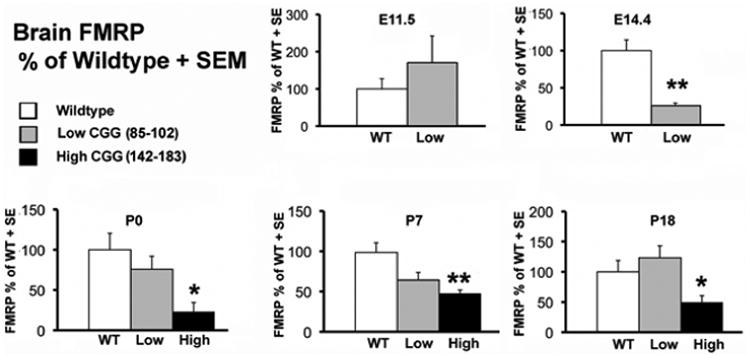
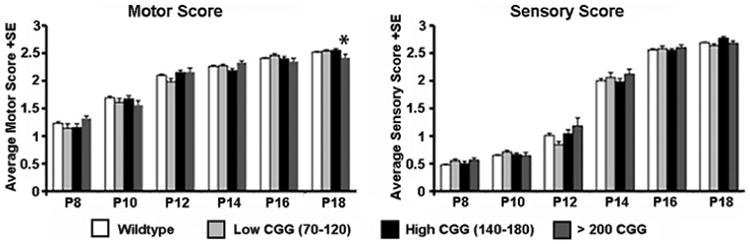
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder that affects some carriers of the fragile X premutation (PM). In PM carriers, there is a moderate expansion of a CGG trinucleotide sequence (55-200 repeats) in the fragile X gene (FMR1) leading to increased FMR1 mRNA and small to moderate decreases in the fragile X mental retardation protein (FMRP) expression. The key symptoms of FXTAS include cerebellar gait ataxia, kinetic tremor, sensorimotor deficits, neuropsychiatric changes, and dementia. While the specific trigger(s) that causes PM carriers to progress to FXTAS pathogenesis remains elusive, the use of animal models has shed light on the underlying neurobiology of the altered pathways involved in disease development. In this review, we examine the current use of mouse models to study PM and FXTAS, focusing on recent advances in the field. Specifically, we will discuss the construct, face, and predictive validities of these PM mouse models, the insights into the underlying disease mechanisms, and potential treatments.
Keywords: CGG trinucleotide repeat; Fragile X mental retardation (FMR1) gene; Fragile X premutation; Fragile X-associated tremor/ataxia syndrome (FXTAS); Mouse models; Neurodegenerative disorder.
Conflict of interest statement
Compliance with Ethical Standards: Conflict of Interest: The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Characterization and Early Detection of Balance Deficits in Fragile X Premutation Carriers With and Without Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS).Cerebellum. 2015 Dec;14(6):650-62. doi: 10.1007/s12311-015-0659-7. Cerebellum. 2015. PMID: 25763861
-
Fragile X-associated tremor/ataxia syndrome: influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles.JAMA Neurol. 2013 Aug;70(8):1022-9. doi: 10.1001/jamaneurol.2013.2934. JAMA Neurol. 2013. PMID: 23753897 Free PMC article.
-
Static and dynamic postural control deficits in aging fragile X mental retardation 1 (FMR1) gene premutation carriers.J Neurodev Disord. 2019 Jan 21;11(1):2. doi: 10.1186/s11689-018-9261-x. J Neurodev Disord. 2019. PMID: 30665341 Free PMC article.
-
Fragile X-associated tremor/ataxia syndrome.Ann N Y Acad Sci. 2015 Mar;1338(1):58-70. doi: 10.1111/nyas.12693. Epub 2015 Jan 26. Ann N Y Acad Sci. 2015. PMID: 25622649 Free PMC article. Review.
-
A Chinese case of fragile X-associated tremor/ataxia syndrome (FXTAS) with orthostatic tremor:case report and literature review on tremor in FXTAS.BMC Neurol. 2020 Apr 20;20(1):145. doi: 10.1186/s12883-020-01726-z. BMC Neurol. 2020. PMID: 32312236 Free PMC article. Review.
Cited by
-
RNA toxicity and foci formation in microsatellite expansion diseases.Curr Opin Genet Dev. 2017 Jun;44:17-29. doi: 10.1016/j.gde.2017.01.005. Epub 2017 Feb 14. Curr Opin Genet Dev. 2017. PMID: 28208060 Free PMC article. Review.
-
Intellectual disability: dendritic anomalies and emerging genetic perspectives.Acta Neuropathol. 2021 Feb;141(2):139-158. doi: 10.1007/s00401-020-02244-5. Epub 2020 Nov 23. Acta Neuropathol. 2021. PMID: 33226471 Free PMC article. Review.
-
General Anesthetic Use in Fragile X Spectrum Disorders.J Neurosurg Anesthesiol. 2019 Jul;31(3):285-290. doi: 10.1097/ANA.0000000000000508. J Neurosurg Anesthesiol. 2019. PMID: 29734272 Free PMC article. Review.
-
In silico, in vitro, and in vivo Approaches to Identify Molecular Players in Fragile X Tremor and Ataxia Syndrome.Front Mol Biosci. 2020 Mar 11;7:31. doi: 10.3389/fmolb.2020.00031. eCollection 2020. Front Mol Biosci. 2020. PMID: 32219099 Free PMC article. Review.
-
Selective rescue of heightened anxiety but not gait ataxia in a premutation 90CGG mouse model of Fragile X-associated tremor/ataxia syndrome.Hum Mol Genet. 2017 Jun 1;26(11):2133-2145. doi: 10.1093/hmg/ddx108. Hum Mol Genet. 2017. PMID: 28369393 Free PMC article.
References
-
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6(5):376–87. - PubMed
-
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–58. - PubMed
-
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14. - PubMed
-
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–30. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
