

Epigenetic Determinants of Cancer
- PMID: 27194046
- PMCID: PMC5008069
- DOI: 10.1101/cshperspect.a019505
Epigenetic Determinants of Cancer
Abstract
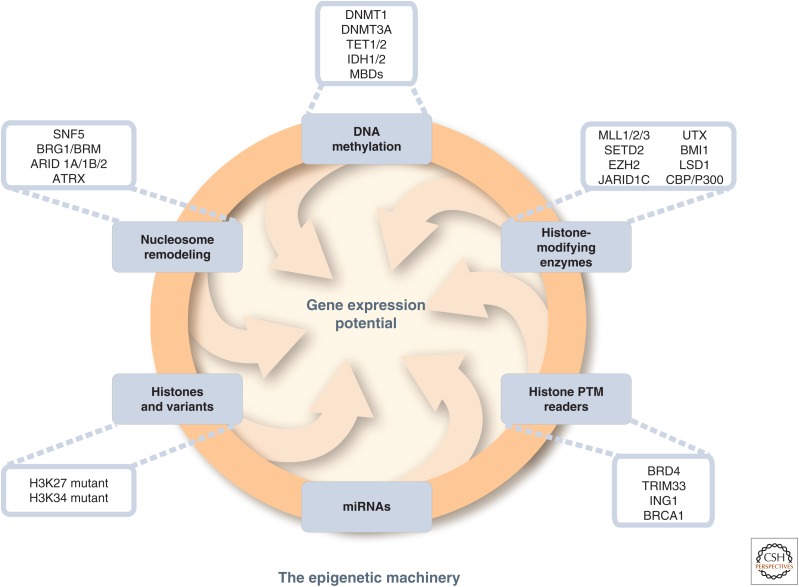
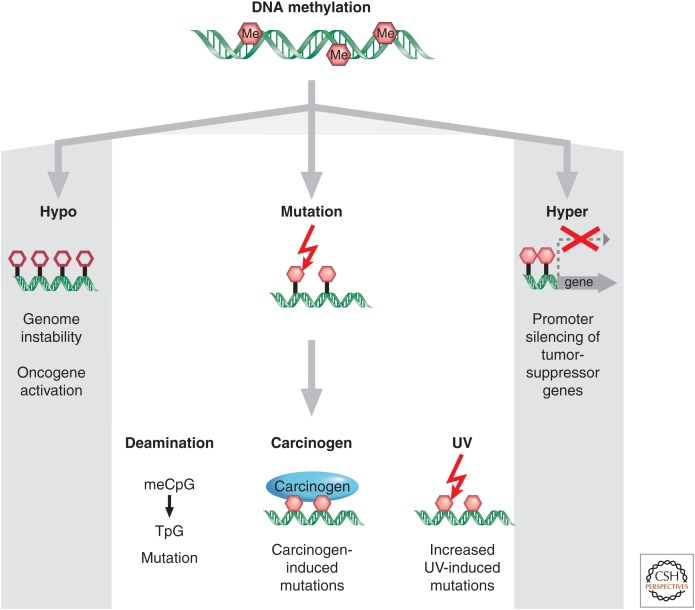
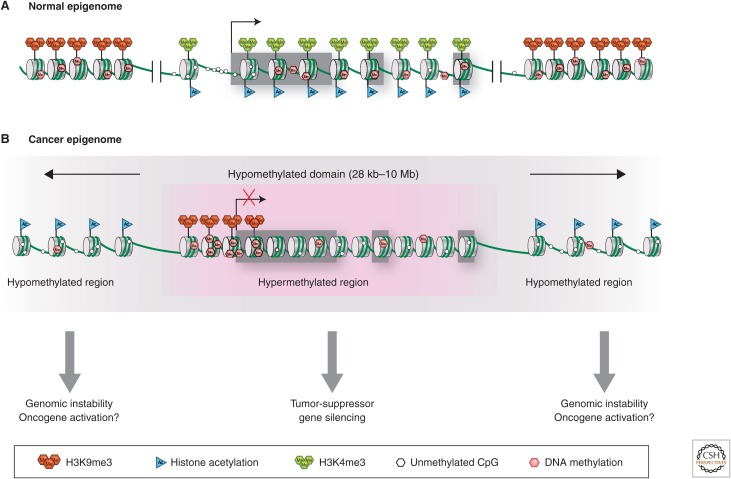
SUMMARYEpigenetic changes are present in all human cancers and are now known to cooperate with genetic alterations to drive the cancer phenotype. These changes involve DNA methylation, histone modifiers and readers, chromatin remodelers, microRNAs, and other components of chromatin. Cancer genetics and epigenetics are inextricably linked in generating the malignant phenotype; epigenetic changes can cause mutations in genes, and, conversely, mutations are frequently observed in genes that modify the epigenome. Epigenetic therapies, in which the goal is to reverse these changes, are now one standard of care for a preleukemic disorder and form of lymphoma. The application of epigenetic therapies in the treatment of solid tumors is also emerging as a viable therapeutic route.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
Similar articles
-
Molecular and Cellular Changes During Cancer Progression Resulting From Genetic and Epigenetic Alterations.Prog Mol Biol Transl Sci. 2016;144:3-47. doi: 10.1016/bs.pmbts.2016.09.001. Epub 2016 Oct 7. Prog Mol Biol Transl Sci. 2016. PMID: 27865461 Review.
-
Epigenetic changes in cancer and preneoplasia.Cold Spring Harb Symp Quant Biol. 2005;70:329-33. doi: 10.1101/sqb.2005.70.036. Cold Spring Harb Symp Quant Biol. 2005. PMID: 16869769 Review.
-
DNA methylation aberrancies as a guide for surveillance and treatment of human cancers.Epigenetics. 2017 Jun 3;12(6):416-432. doi: 10.1080/15592294.2017.1311434. Epub 2017 Mar 30. Epigenetics. 2017. PMID: 28358281 Free PMC article. Review.
-
[DNA methylation and cancer].Gan To Kagaku Ryoho. 2007 Feb;34(2):145-9. Gan To Kagaku Ryoho. 2007. PMID: 17301518 Review. Japanese.
-
Epigenetic changes in cancer.APMIS. 2007 Oct;115(10):1039-59. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x. APMIS. 2007. PMID: 18042143 Review.
Cited by
-
Therapeutic modulation of gene expression in the disease state: Treatment strategies and approaches for the development of next-generation of the epigenetic drugs.Front Bioeng Biotechnol. 2022 Oct 17;10:1035543. doi: 10.3389/fbioe.2022.1035543. eCollection 2022. Front Bioeng Biotechnol. 2022. PMID: 36324900 Free PMC article. Review.
-
A prognostic 11-DNA methylation signature for lung squamous cell carcinoma.J Thorac Dis. 2020 May;12(5):2569-2582. doi: 10.21037/jtd.2020.03.31. J Thorac Dis. 2020. PMID: 32642165 Free PMC article.
-
Epigenetic priming targets tumor heterogeneity to shift transcriptomic phenotype of pancreatic ductal adenocarcinoma towards a Vitamin D susceptible state.Cell Death Dis. 2024 Jan 26;15(1):89. doi: 10.1038/s41419-024-06460-9. Cell Death Dis. 2024. PMID: 38272889 Free PMC article.
-
Histone Acyl Code in Precision Oncology: Mechanistic Insights from Dietary and Metabolic Factors.Nutrients. 2024 Jan 30;16(3):396. doi: 10.3390/nu16030396. Nutrients. 2024. PMID: 38337680 Free PMC article. Review.
-
The Anticancer Potential of Plant-Derived Nutraceuticals via the Modulation of Gene Expression.Plants (Basel). 2022 Sep 26;11(19):2524. doi: 10.3390/plants11192524. Plants (Basel). 2022. PMID: 36235389 Free PMC article. Review.
References
-
- Allis CD, Jenuwein T, Reinberg D. 2014. Overview and concepts. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a018739. - DOI
-
- Araki S, Doi H, Sano Y, Tanaka S, Miyake Y. 2009. Preparation and CO(2) adsorption properties of aminopropyl-functionalized mesoporous silica microspheres. J Colloid Interface Sci 339: 382–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases