

Derivatives of 6-cinnamamido-quinoline-4-carboxamide impair lysosome function and induce apoptosis
- PMID: 27191263
- PMCID: PMC5122373
- DOI: 10.18632/oncotarget.9348
Derivatives of 6-cinnamamido-quinoline-4-carboxamide impair lysosome function and induce apoptosis
Abstract
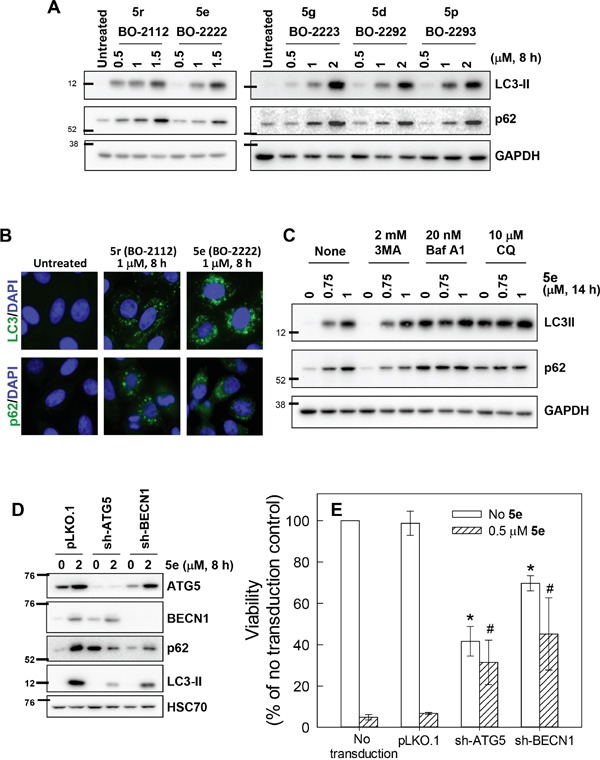
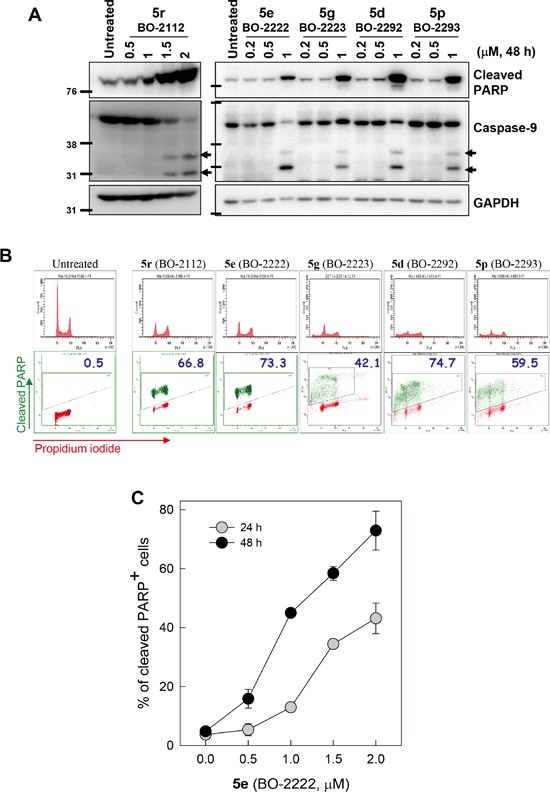
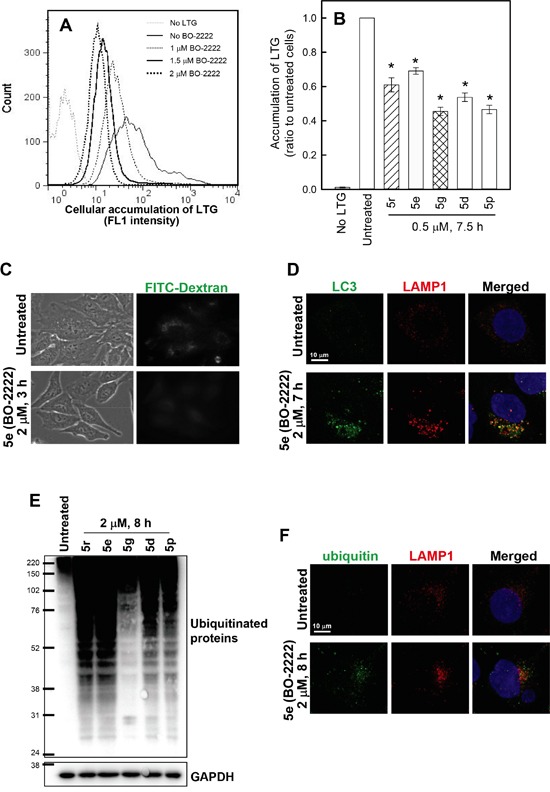
Autophagy is a lysosomal degradative process that protects cancer cells from multiple types of stress. In this study, we synthesized a series of derivatives of 6-cinnamamido-quinoline-4-carboxamide (CiQ), and investigated their effects on the proliferation and autophagy of cancer cells in vitro. These derivatives effectively inhibited the proliferation of a broad spectrum of cancer cell lines. Further study revealed that CiQ derivatives may induce autophagy and result in disruption of autophagy propagation. Consequently, these derivatives triggered massive apoptosis, as evidenced by caspase-9 activation and PARP cleavage. Blockage of autophagy by depletion of autophagy related gene ATG5 or BECN1 considerably alleviated CiQ-induced cell death, indicating that autophagy may mediate CiQ-induced cell death. Furthermore, treatment with CiQ derivatives increased lysosome membrane permeability (LMP) and enhanced accumulation of ubiquitinated proteins, which collectively indicate impaired lysosome function. In addition, treatment of cells with CiQ derivatives activated extracellular signal-regulated kinase (ERK); abrogation of ERK activation, either by treating cells with U0126, an inhibitor of mitogen-activated protein/ERK kinase 1 (MEK1), or by ectopically overexpressing a dominant-negative MEK1, significantly reduced CiQ derivative-induced LMP, LC3 and p62 accumulation, and cytotoxicity. These results indicate that CiQ derivatives activate ERK and disrupt lysosome function, thereby altering autophagic flux and resulting in apoptotic cell death.
Keywords: ERK; anticancer; autophagy; cinnamamide quinolines; lysosome.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures



Similar articles
-
The dual PI3K/mTOR inhibitor NVP-BEZ235 and chloroquine synergize to trigger apoptosis via mitochondrial-lysosomal cross-talk.Int J Cancer. 2013 Jun 1;132(11):2682-93. doi: 10.1002/ijc.27935. Epub 2012 Dec 4. Int J Cancer. 2013. PMID: 23151917
-
Hispidin induces autophagic and necrotic death in SGC-7901 gastric cancer cells through lysosomal membrane permeabilization by inhibiting tubulin polymerization.Oncotarget. 2017 Apr 18;8(16):26992-27006. doi: 10.18632/oncotarget.15935. Oncotarget. 2017. PMID: 28460485 Free PMC article.
-
6-Hydroxydopamine induces autophagic flux dysfunction by impairing transcription factor EB activation and lysosomal function in dopaminergic neurons and SH-SY5Y cells.Toxicol Lett. 2018 Feb;283:58-68. doi: 10.1016/j.toxlet.2017.11.017. Epub 2017 Nov 21. Toxicol Lett. 2018. PMID: 29170033
-
Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells.Autophagy. 2012 Mar;8(3):338-49. doi: 10.4161/auto.18721. Epub 2012 Feb 3. Autophagy. 2012. PMID: 22302005
-
The Achilles' heel of cancer: targeting tumors via lysosome-induced immunogenic cell death.Cell Death Dis. 2022 May 30;13(5):509. doi: 10.1038/s41419-022-04912-8. Cell Death Dis. 2022. PMID: 35637197 Free PMC article. Review.
Cited by
-
Targeting Tristetraprolin Expression or Functional Activity Regulates Inflammatory Response Induced by MSU Crystals.Front Immunol. 2021 Jul 16;12:675534. doi: 10.3389/fimmu.2021.675534. eCollection 2021. Front Immunol. 2021. PMID: 34335573 Free PMC article.
-
Demethylzeylasteral (T-96) initiates extrinsic apoptosis against prostate cancer cells by inducing ROS-mediated ER stress and suppressing autophagic flux.Biol Res. 2021 Sep 6;54(1):27. doi: 10.1186/s40659-021-00350-6. Biol Res. 2021. PMID: 34488902 Free PMC article.
-
Autophagic flux disruption contributes to Ganoderma lucidum polysaccharide-induced apoptosis in human colorectal cancer cells via MAPK/ERK activation.Cell Death Dis. 2019 Jun 11;10(6):456. doi: 10.1038/s41419-019-1653-7. Cell Death Dis. 2019. PMID: 31186406 Free PMC article.
-
Role of lysosomes in physiological activities, diseases, and therapy.J Hematol Oncol. 2021 May 14;14(1):79. doi: 10.1186/s13045-021-01087-1. J Hematol Oncol. 2021. PMID: 33990205 Free PMC article. Review.
-
ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer.Cells. 2021 Sep 22;10(10):2509. doi: 10.3390/cells10102509. Cells. 2021. PMID: 34685488 Free PMC article. Review.
References
-
- Homewood CA, Warhurst DC, Peters W, Baggaley VC. Lysosomes, pH and the anti-malarial action of chloroquine. Nature. 1972;235:50–52. - PubMed
-
- Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220–233. - PubMed
-
- Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, Sun H, Wang X, Skrtic M, Seneviratne A, Cusimano M, Jhas B, Gronda M, MacLean N, Cho EE, Spagnuolo PA, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. 2013;123:315–328. - PMC - PubMed
-
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8258. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
