

Retinoblastoma
- PMID: 27189421
- PMCID: PMC5744255
- DOI: 10.1038/nrdp.2015.21
Retinoblastoma
Abstract
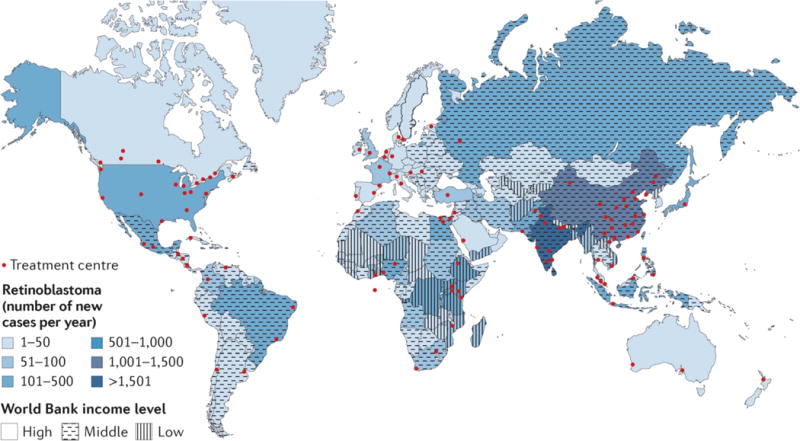
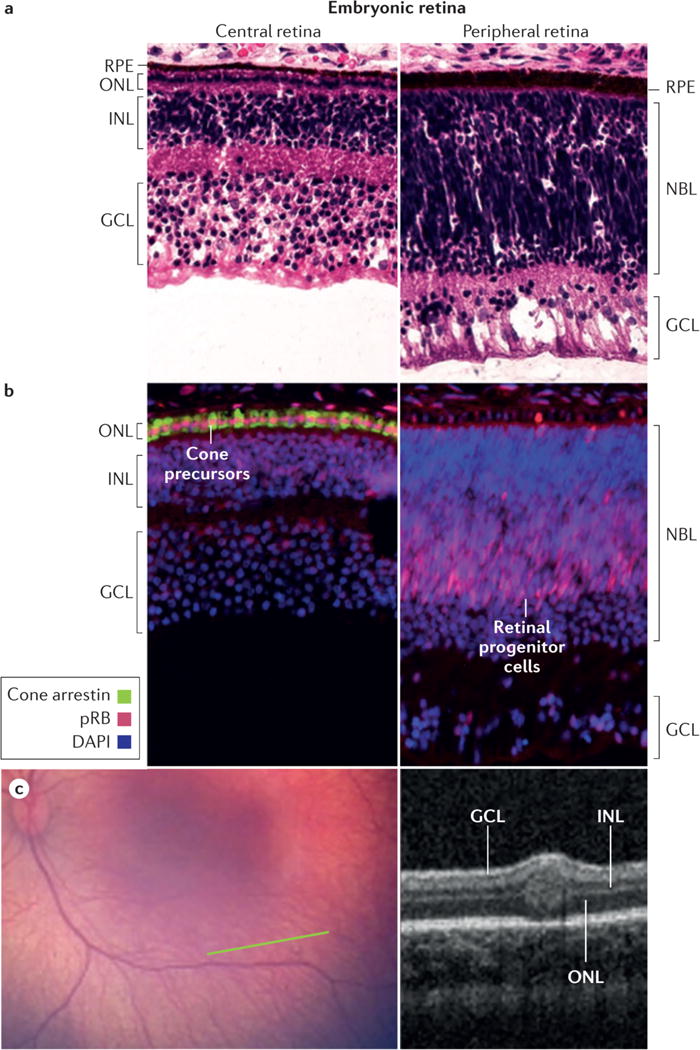
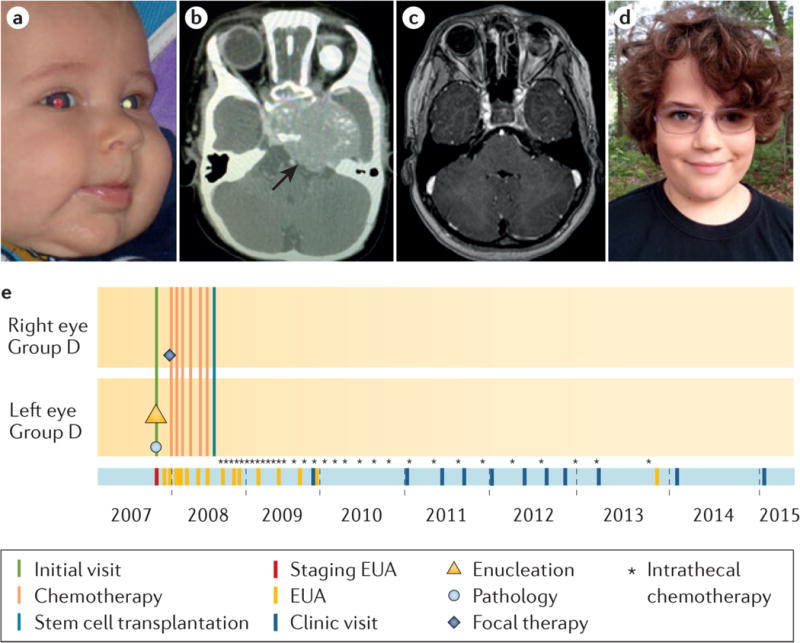
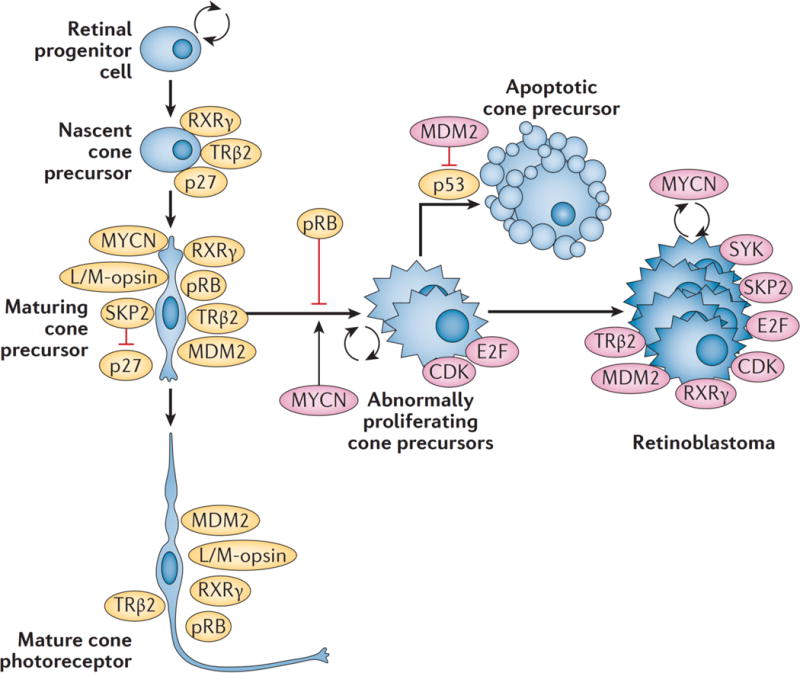
Retinoblastoma is a rare cancer of the infant retina that is diagnosed in approximately 8,000 children each year worldwide. It forms when both retinoblastoma gene (RB1) alleles are mutated in a susceptible retinal cell, probably a cone photoreceptor precursor. Loss of the tumour-suppressive functions of the retinoblastoma protein (pRB) leads to uncontrolled cell division and recurrent genomic changes during tumour progression. Although pRB is expressed in almost all tissues, cone precursors have biochemical and molecular features that may sensitize them to RB1 loss and enable tumorigenesis. Patient survival is >95% in high-income countries but <30% globally. However, outcomes are improving owing to increased disease awareness for earlier diagnosis, application of new guidelines and sharing of expertise. Intra-arterial and intravitreal chemotherapy have emerged as promising methods to salvage eyes that with conventional treatment might have been lost. Ongoing international collaborations will replace the multiple different classifications of eye involvement with standardized definitions to consistently assess the eligibility, efficacy and safety of treatment options. Life-long follow-up is warranted, as survivors of heritable retinoblastoma are at risk for developing second cancers. Defining the molecular consequences of RB1 loss in diverse tissues may open new avenues for treatment and prevention of retinoblastoma, as well as second cancers, in patients with germline RB1 mutations.
Conflict of interest statement
There is NO competing interest.
Figures



Similar articles
-
Novel germline mutation in the RB1 gene with multifocal bone tumors following retinoblastoma.Pediatr Int. 2003 Dec;45(6):728-30. doi: 10.1111/j.1442-200x.2003.01821.x. Pediatr Int. 2003. PMID: 14651550 No abstract available.
-
RB1 mutations and second primary malignancies after hereditary retinoblastoma.Fam Cancer. 2012 Jun;11(2):225-33. doi: 10.1007/s10689-011-9505-3. Fam Cancer. 2012. PMID: 22205104 Free PMC article.
-
Uncommon RB1 somatic mutations in a unilateral retinoblastoma patient.Medicina (B Aires). 2015;75(3):137-41. Medicina (B Aires). 2015. PMID: 26117602
-
Retinoblastoma: the disease, gene and protein provide critical leads to understand cancer.Semin Cancer Biol. 2000 Aug;10(4):255-69. doi: 10.1006/scbi.2000.0326. Semin Cancer Biol. 2000. PMID: 10966849 Review.
-
[From gene to disease; retinoblastoma and the RB1 gene].Ned Tijdschr Geneeskd. 2001 Jun 30;145(26):1245-7. Ned Tijdschr Geneeskd. 2001. PMID: 11455690 Review. Dutch.
Cited by
-
Stereotactic Gamma Knife® Radiosurgery of Intraocular Retinoblastoma: Six-Year Experience.Cureus. 2022 Sep 3;14(9):e28751. doi: 10.7759/cureus.28751. eCollection 2022 Sep. Cureus. 2022. PMID: 36211113 Free PMC article.
-
Retinoblastoma tumor cell proliferation is negatively associated with an immune gene expression signature and increased immune cells.Lab Invest. 2021 Jun;101(6):701-718. doi: 10.1038/s41374-021-00573-x. Epub 2021 Mar 3. Lab Invest. 2021. PMID: 33658609
-
Long non-coding RNA CASC9 promotes the progression of retinoblastoma via interacting with miR-145-5p.Cell Cycle. 2020 Sep;19(18):2270-2280. doi: 10.1080/15384101.2020.1802813. Epub 2020 Aug 10. Cell Cycle. 2020. PMID: 32772636 Free PMC article.
-
Treatment Outcome of Children with Retinoblastoma in a Tertiary Care Referral Hospital in Indonesia.Asian Pac J Cancer Prev. 2021 May 1;22(5):1613-1621. doi: 10.31557/APJCP.2021.22.5.1613. Asian Pac J Cancer Prev. 2021. PMID: 34048193 Free PMC article.
-
Feasibility of Proton Beam Therapy as a Rescue Therapy in Heavily Pre-Treated Retinoblastoma Eyes.Cancers (Basel). 2021 Apr 13;13(8):1862. doi: 10.3390/cancers13081862. Cancers (Basel). 2021. PMID: 33924716 Free PMC article.
References
-
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. - PubMed
-
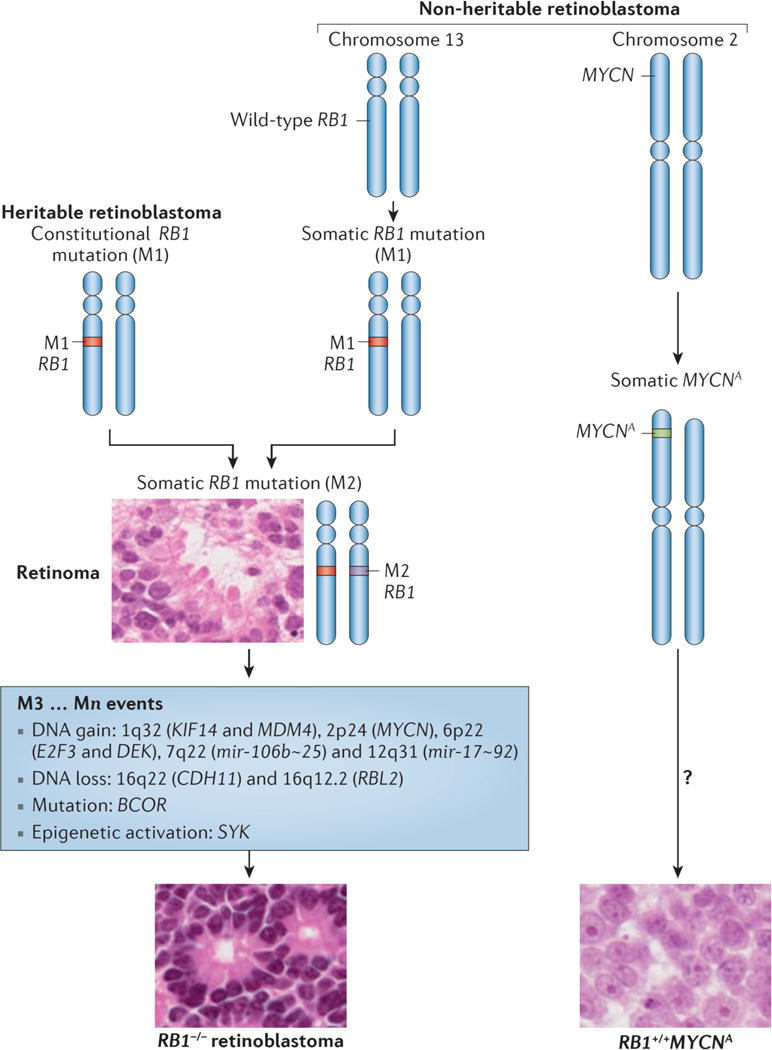
- Dimaras H, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363–1372. - PubMed
-
- Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93:1129–1131. doi:93/9/1129 [pii]10.1136/bjo.2008.150292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
