

mCAL: A New Approach for Versatile Multiplex Action of Cas9 Using One sgRNA and Loci Flanked by a Programmed Target Sequence
- PMID: 27185399
- PMCID: PMC4938667
- DOI: 10.1534/g3.116.029801
mCAL: A New Approach for Versatile Multiplex Action of Cas9 Using One sgRNA and Loci Flanked by a Programmed Target Sequence
Erratum in
-
Corrigendum.G3 (Bethesda). 2019 Apr 9;9(4):1279. doi: 10.1534/g3.119.400096. G3 (Bethesda). 2019. PMID: 30967430 Free PMC article. No abstract available.
Abstract
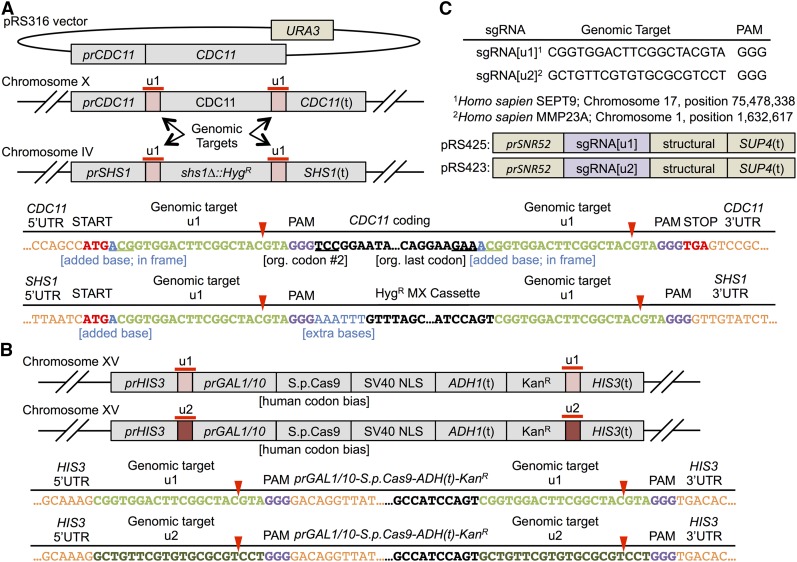
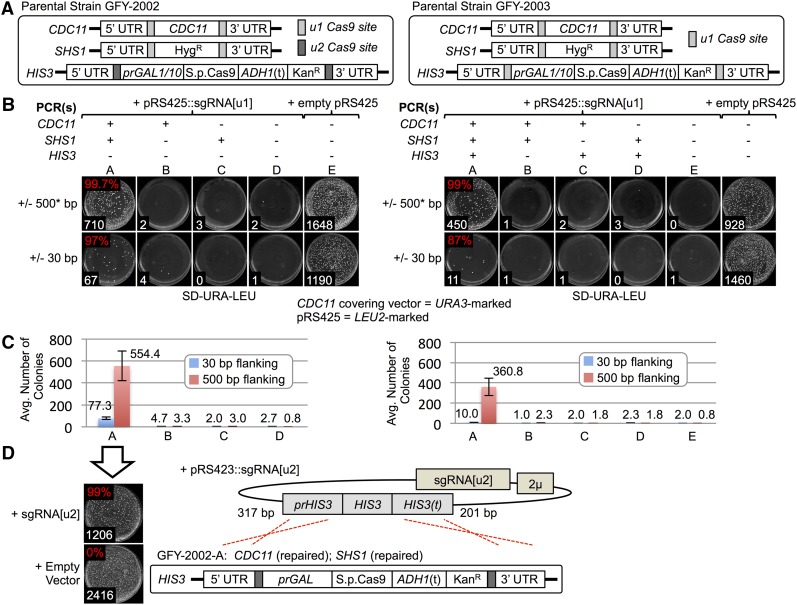
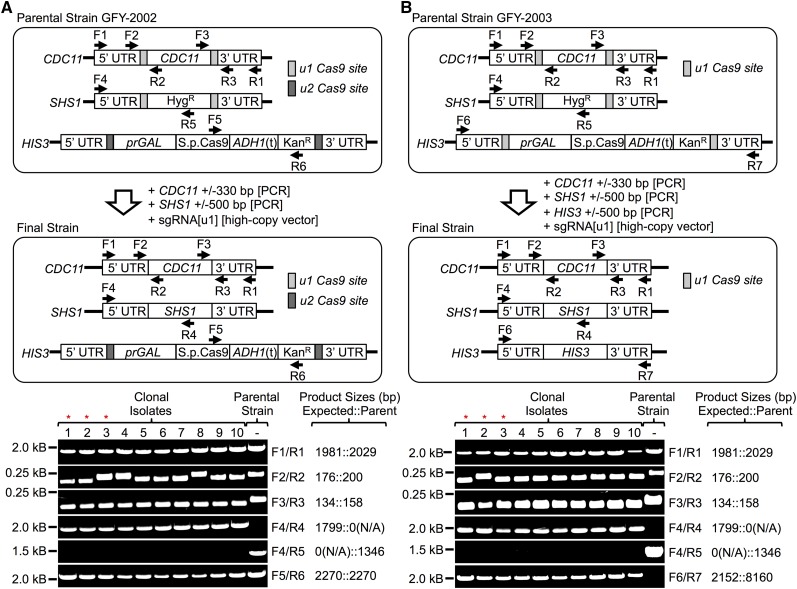
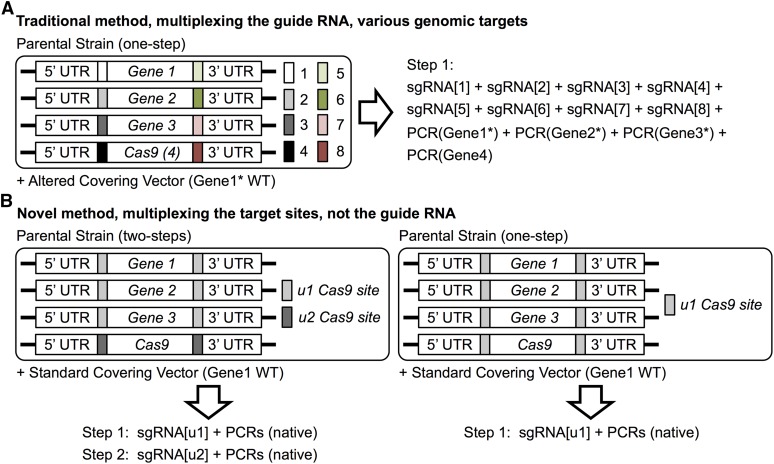
Genome editing exploiting CRISPR/Cas9 has been adopted widely in academia and in the biotechnology industry to manipulate DNA sequences in diverse organisms. Molecular engineering of Cas9 itself and its guide RNA, and the strategies for using them, have increased efficiency, optimized specificity, reduced inappropriate off-target effects, and introduced modifications for performing other functions (transcriptional regulation, high-resolution imaging, protein recruitment, and high-throughput screening). Moreover, Cas9 has the ability to multiplex, i.e., to act at different genomic targets within the same nucleus. Currently, however, introducing concurrent changes at multiple loci involves: (i) identification of appropriate genomic sites, especially the availability of suitable PAM sequences; (ii) the design, construction, and expression of multiple sgRNA directed against those sites; (iii) potential difficulties in altering essential genes; and (iv) lingering concerns about "off-target" effects. We have devised a new approach that circumvents these drawbacks, as we demonstrate here using the yeast Saccharomyces cerevisiae First, any gene(s) of interest are flanked upstream and downstream with a single unique target sequence that does not normally exist in the genome. Thereafter, expression of one sgRNA and cotransformation with appropriate PCR fragments permits concomitant Cas9-mediated alteration of multiple genes (both essential and nonessential). The system we developed also allows for maintenance of the integrated, inducible Cas9-expression cassette or its simultaneous scarless excision. Our scheme-dubbed mCAL for " M: ultiplexing of C: as9 at A: rtificial L: oci"-can be applied to any organism in which the CRISPR/Cas9 methodology is currently being utilized. In principle, it can be applied to install synthetic sequences into the genome, to generate genomic libraries, and to program strains or cell lines so that they can be conveniently (and repeatedly) manipulated at multiple loci with extremely high efficiency.
Keywords: CRISPR; Saccharomyces cerevisiae; essential genes; genome editing; genome engineering.
Copyright © 2016 Finnigan and Thorner.
Figures
Similar articles
-
[CRISPR/CAS9, the King of Genome Editing Tools].Mol Biol (Mosk). 2017 Jul-Aug;51(4):582-594. doi: 10.7868/S0026898417040036. Mol Biol (Mosk). 2017. PMID: 28900076 Review. Russian.
-
Optimization of genome editing through CRISPR-Cas9 engineering.Bioengineered. 2016 Apr;7(3):166-74. doi: 10.1080/21655979.2016.1189039. Bioengineered. 2016. PMID: 27340770 Free PMC article. Review.
-
CRISPR/Cas9 in Genome Editing and Beyond.Annu Rev Biochem. 2016 Jun 2;85:227-64. doi: 10.1146/annurev-biochem-060815-014607. Epub 2016 Apr 25. Annu Rev Biochem. 2016. PMID: 27145843 Review.
-
Guide RNA engineering for versatile Cas9 functionality.Nucleic Acids Res. 2016 Nov 16;44(20):9555-9564. doi: 10.1093/nar/gkw908. Epub 2016 Oct 12. Nucleic Acids Res. 2016. PMID: 27733506 Free PMC article. Review.
-
Precision genome editing in the CRISPR era.Biochem Cell Biol. 2017 Apr;95(2):187-201. doi: 10.1139/bcb-2016-0137. Epub 2016 Sep 29. Biochem Cell Biol. 2017. PMID: 28177771 Review.
Cited by
-
CATS: Cas9-assisted tag switching. A high-throughput method for exchanging genomic peptide tags in yeast.BMC Genomics. 2020 Mar 10;21(1):221. doi: 10.1186/s12864-020-6634-9. BMC Genomics. 2020. PMID: 32156257 Free PMC article.
-
Phosphorylation of mRNA-Binding Proteins Puf1 and Puf2 by TORC2-Activated Protein Kinase Ypk1 Alleviates Their Repressive Effects.Membranes (Basel). 2021 Jun 30;11(7):500. doi: 10.3390/membranes11070500. Membranes (Basel). 2021. PMID: 34209236 Free PMC article.
-
Phosphorylation by the stress-activated MAPK Slt2 down-regulates the yeast TOR complex 2.Genes Dev. 2018 Dec 1;32(23-24):1576-1590. doi: 10.1101/gad.318709.118. Epub 2018 Nov 26. Genes Dev. 2018. PMID: 30478248 Free PMC article.
-
Yeast genetic interaction screens in the age of CRISPR/Cas.Curr Genet. 2019 Apr;65(2):307-327. doi: 10.1007/s00294-018-0887-8. Epub 2018 Sep 25. Curr Genet. 2019. PMID: 30255296 Free PMC article. Review.
-
Multiplex Genome Editing in Yeast by CRISPR/Cas9 - A Potent and Agile Tool to Reconstruct Complex Metabolic Pathways.Front Plant Sci. 2021 Aug 5;12:719148. doi: 10.3389/fpls.2021.719148. eCollection 2021. Front Plant Sci. 2021. PMID: 34421973 Free PMC article. Review.
References
-
- Amberg, D. C., D. J. Burke, and J. N. Strathern, 2006 2006 Yeast DNA isolation: miniprep, CSH Protoc. DOI: 10.1101/pdb.prot4148. - PubMed
-
- Bao Z., Xiao H., Liang J., Zhang L., Xiong X., et al. , 2015. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 4: 585–594. - PubMed
-
- Boeke J. D., LaCroute F., Fink G. R., 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197: 345–346. - PubMed
-
- Bolukbasi M. F., Gupta A., Wolfe S. A., 2015. Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat. Methods 13: 41–50. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous