

The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development
- PMID: 27180008
- PMCID: PMC4914454
- DOI: 10.1016/j.phrs.2016.05.008
The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development
Abstract
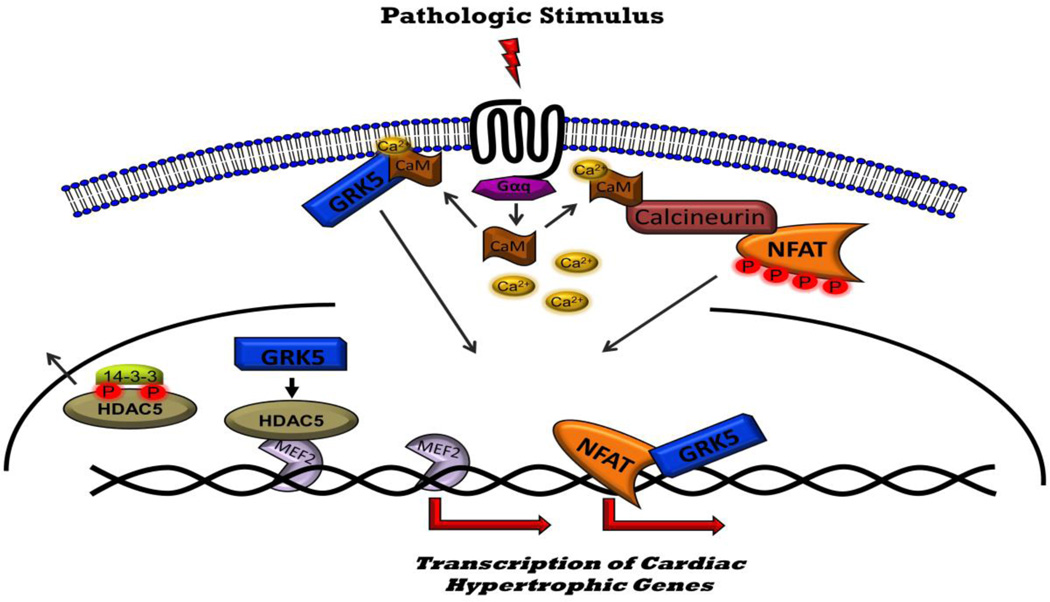
Heart failure (HF) is a global epidemic with the highest degree of mortality and morbidity of any disease presently studied. G protein-coupled receptors (GPCRs) are prominent regulators of cardiovascular function. Activated GPCRs are "turned off" by GPCR kinases (GRKs) in a process known as "desensitization". GRKs 2 and 5 are highly expressed in the heart, and known to be upregulated in HF. Over the last 20 years, both GRK2 and GRK5 have been demonstrated to be critical mediators of the molecular alterations that occur in the failing heart. In the present review, we will highlight recent findings that further characterize "non-canonical" GRK signaling observed in HF. Further, we will also present potential therapeutic strategies (i.e. small molecule inhibition, microRNAs, gene therapy) that may have potential in combating the deleterious effects of GRKs in HF.
Keywords: G protein-coupled receptor; G protein-coupled receptor kinase; Heart failure; Hypertrophy; Myocardium.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
G protein-coupled receptor kinases in normal and failing myocardium.Front Biosci (Landmark Ed). 2011 Jun 1;16(8):3047-60. doi: 10.2741/3898. Front Biosci (Landmark Ed). 2011. PMID: 21622221 Free PMC article. Review.
-
"Canonical and non-canonical actions of GRK5 in the heart".J Mol Cell Cardiol. 2016 Mar;92:196-202. doi: 10.1016/j.yjmcc.2016.01.027. Epub 2016 Jan 29. J Mol Cell Cardiol. 2016. PMID: 26829117 Free PMC article. Review.
-
Utilizing a structure-based docking approach to develop potent G protein-coupled receptor kinase (GRK) 2 and 5 inhibitors.Bioorg Med Chem Lett. 2018 May 15;28(9):1507-1515. doi: 10.1016/j.bmcl.2018.03.082. Epub 2018 Mar 30. Bioorg Med Chem Lett. 2018. PMID: 29627263 Free PMC article.
-
Antagonistic Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Reveal GRK5-Mediated Cardioprotection via Mineralocorticoid Receptor Inhibition.Int J Mol Sci. 2020 Apr 20;21(8):2868. doi: 10.3390/ijms21082868. Int J Mol Sci. 2020. PMID: 32326036 Free PMC article.
-
Novel roles for G protein-coupled receptor kinases in cardiac injury and repair.Biochem Soc Trans. 2023 Apr 26;51(2):715-724. doi: 10.1042/BST20221317. Biochem Soc Trans. 2023. PMID: 37013982 Review.
Cited by
-
Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease.Int J Mol Sci. 2018 Nov 27;19(12):3764. doi: 10.3390/ijms19123764. Int J Mol Sci. 2018. PMID: 30486399 Free PMC article. Review.
-
Targeting GRK5 for Treating Chronic Degenerative Diseases.Int J Mol Sci. 2021 Feb 15;22(4):1920. doi: 10.3390/ijms22041920. Int J Mol Sci. 2021. PMID: 33671974 Free PMC article. Review.
-
GRK5 - A Functional Bridge Between Cardiovascular and Neurodegenerative Disorders.Front Pharmacol. 2018 Dec 17;9:1484. doi: 10.3389/fphar.2018.01484. eCollection 2018. Front Pharmacol. 2018. PMID: 30618771 Free PMC article. Review.
-
Comprehensive insights in GRK4 and hypertension: From mechanisms to potential therapeutics.Pharmacol Ther. 2022 Nov;239:108194. doi: 10.1016/j.pharmthera.2022.108194. Epub 2022 Apr 27. Pharmacol Ther. 2022. PMID: 35487286 Free PMC article. Review.
-
Sex/Gender- and Age-Related Differences in β-Adrenergic Receptor Signaling in Cardiovascular Diseases.J Clin Med. 2022 Jul 22;11(15):4280. doi: 10.3390/jcm11154280. J Clin Med. 2022. PMID: 35893368 Free PMC article. Review.
References
-
- Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 2007;190(1):9–19. - PubMed
-
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. - PubMed
-
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2(10):727–733. - PubMed
-
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
