

PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects
- PMID: 27172900
- PMCID: PMC5129924
- DOI: 10.18632/oncotarget.9258
PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects
Abstract
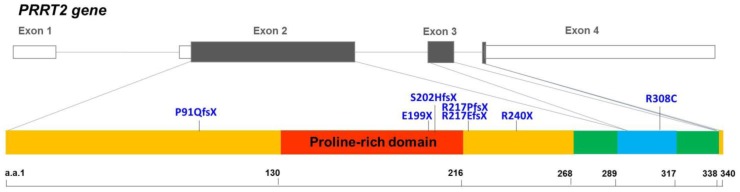
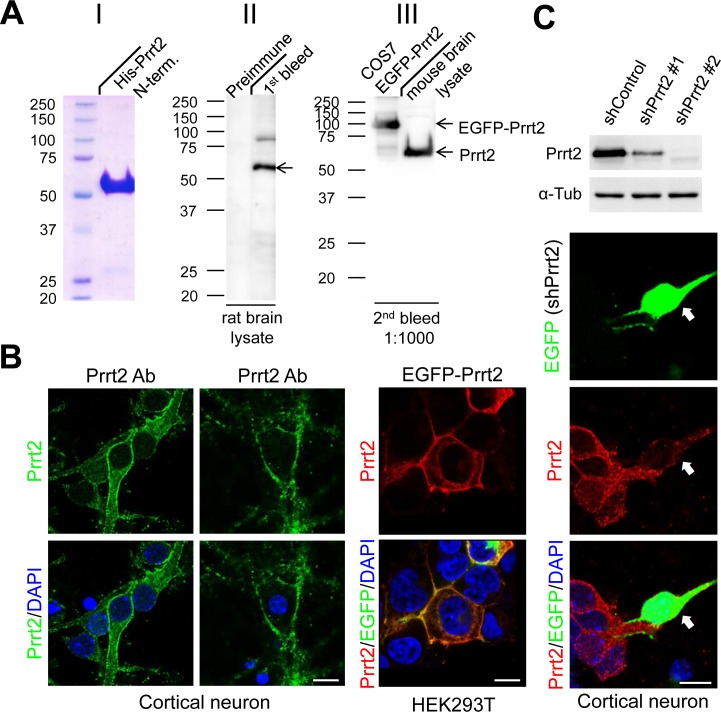
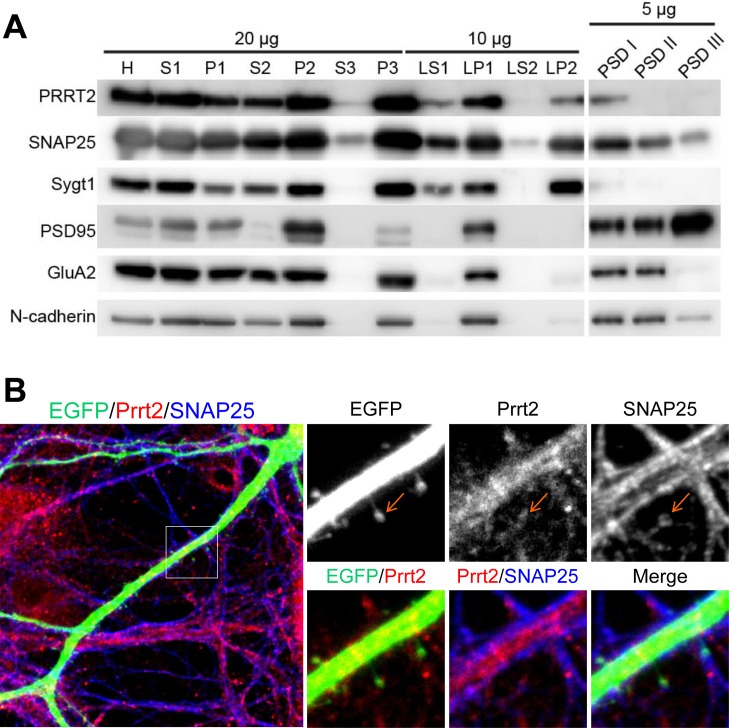
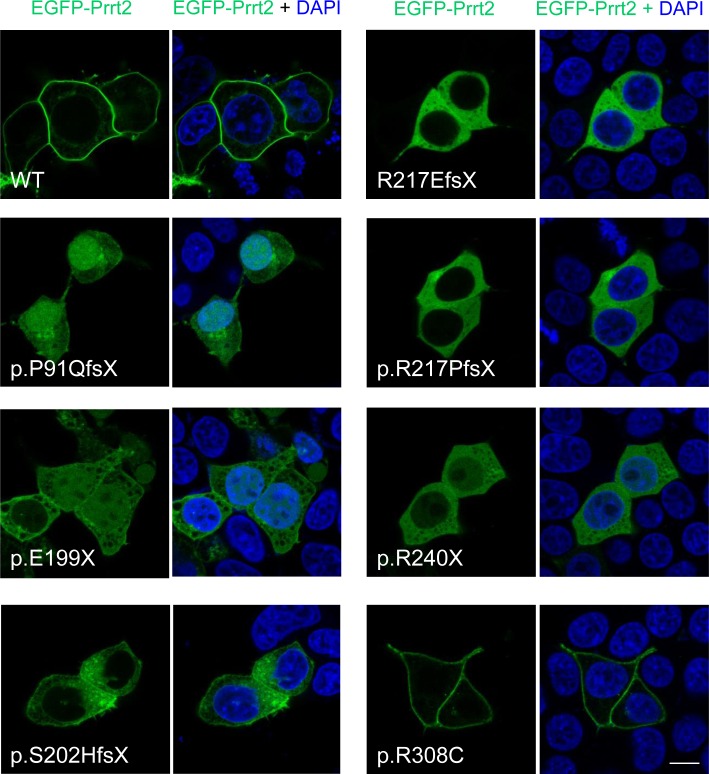
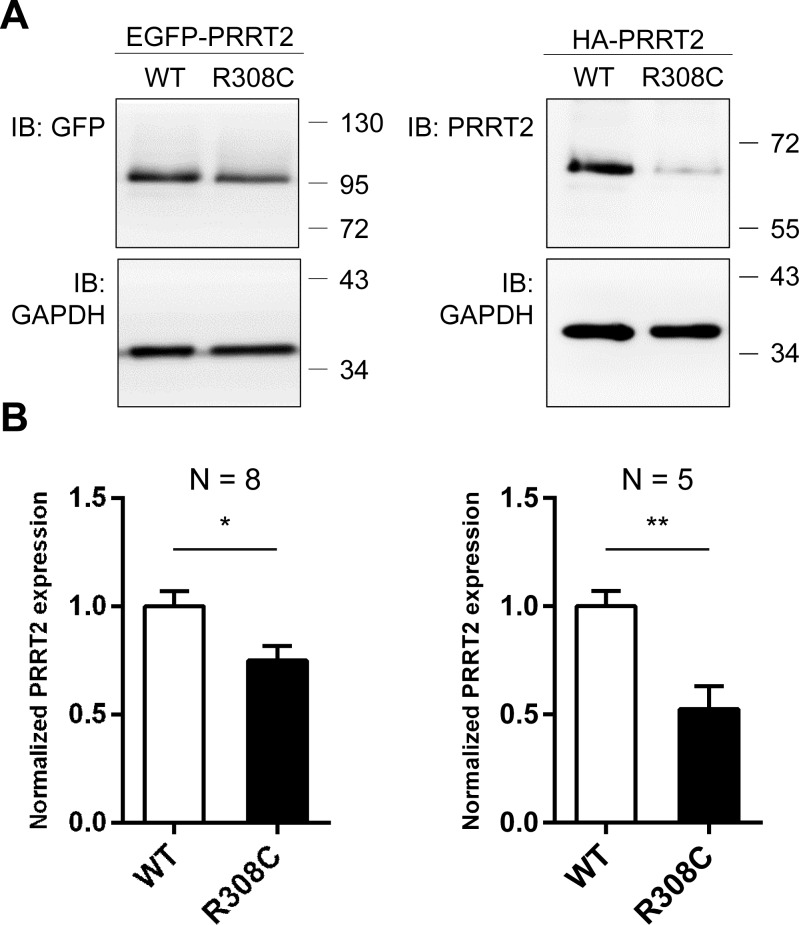
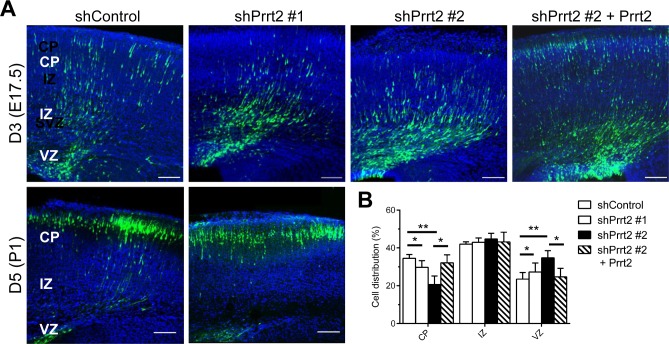
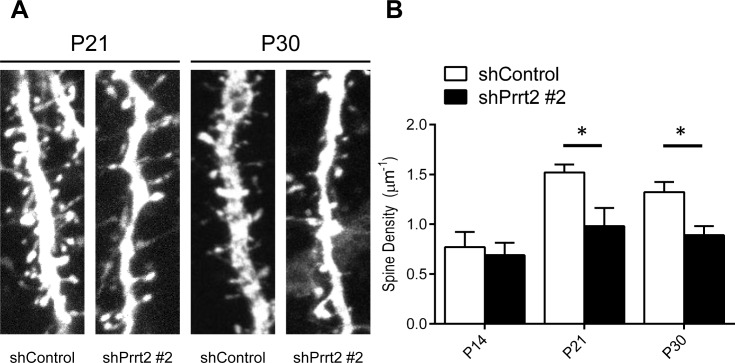
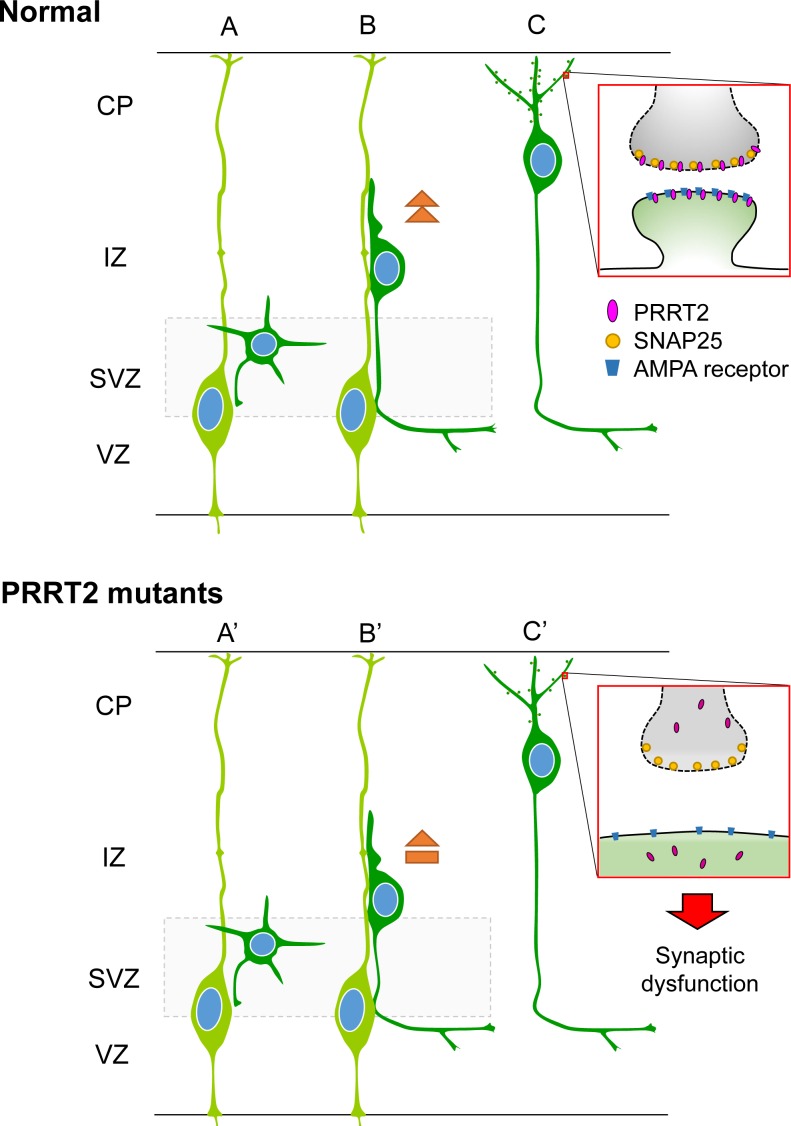
Mutations in the proline-rich transmembrane protein 2 (PRRT2) gene cause a wide spectrum of neurological diseases, ranging from paroxysmal kinesigenic dyskinesia (PKD) to mental retardation and epilepsy. Previously, seven PKD-related PRRT2 heterozygous mutations were identified in the Taiwanese population: P91QfsX, E199X, S202HfsX, R217PfsX, R217EfsX, R240X and R308C. This study aimed to investigate the disease-causing mechanisms of these PRRT2 mutations. We first documented that Prrt2 was localized at the pre- and post-synaptic membranes with a close spatial association with SNAP25 by synaptic membrane fractionation and immunostaining of the rat neurons. Our results then revealed that the six truncating Prrt2 mutants were accumulated in the cytoplasm and thus failed to target to the cell membrane; the R308C missense mutant had significantly reduced protein expression, suggesting loss-of function effects generated by these mutations. Using in utero electroporation of shRNA into cortical neurons, we further found that knocking down Prrt2 expression in vivo resulted in a delay in neuronal migration during embryonic development and a marked decrease in synaptic density after birth. These pathologic effects and novel disease-causing mechanisms may contribute to the severe clinical symptoms in PRRT2-related diseases.
Keywords: PRRT2; Pathology Section; Taiwan; neuronal migration; paroxysmal kinesigenic dyskinesia (PKD); synaptic development.
Conflict of interest statement
All authors declare no conflicts of interest.
Figures
Similar articles
-
PRRT2 missense mutations cluster near C-terminus and frequently lead to protein mislocalization.Epilepsia. 2019 May;60(5):807-817. doi: 10.1111/epi.14725. Epub 2019 Apr 13. Epilepsia. 2019. PMID: 30980674
-
The evolving spectrum of PRRT2-associated paroxysmal diseases.Brain. 2015 Dec;138(Pt 12):3476-95. doi: 10.1093/brain/awv317. Epub 2015 Nov 23. Brain. 2015. PMID: 26598493 Review.
-
Clinical spectrum and genotype-phenotype correlations in PRRT2 Italian patients.Eur J Paediatr Neurol. 2020 Sep;28:193-197. doi: 10.1016/j.ejpn.2020.06.005. Epub 2020 Jun 23. Eur J Paediatr Neurol. 2020. PMID: 32651081
-
PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions.Neurology. 2012 Aug 21;79(8):777-84. doi: 10.1212/WNL.0b013e3182661fe3. Epub 2012 Aug 8. Neurology. 2012. PMID: 22875091
-
PRRT2 mutations in a cohort of Chinese families with paroxysmal kinesigenic dyskinesia and genotype-phenotype correlation reanalysis in literatures.Int J Neurosci. 2018 Aug;128(8):751-760. doi: 10.1080/00207454.2017.1418345. Epub 2018 Jan 7. Int J Neurosci. 2018. PMID: 29285950 Review.
Cited by
-
Age-dependent neurological phenotypes in a mouse model of PRRT2-related diseases.Neurogenetics. 2021 Jul;22(3):171-185. doi: 10.1007/s10048-021-00645-6. Epub 2021 Jun 8. Neurogenetics. 2021. PMID: 34101060 Free PMC article.
-
The Spectrum of PRRT2-Associated Disorders: Update on Clinical Features and Pathophysiology.Front Neurol. 2021 Mar 4;12:629747. doi: 10.3389/fneur.2021.629747. eCollection 2021. Front Neurol. 2021. PMID: 33746883 Free PMC article. Review.
-
Presynaptic PRRT2 Deficiency Causes Cerebellar Dysfunction and Paroxysmal Kinesigenic Dyskinesia.Neuroscience. 2020 Nov 10;448:272-286. doi: 10.1016/j.neuroscience.2020.08.034. Epub 2020 Sep 4. Neuroscience. 2020. PMID: 32891704 Free PMC article.
-
Aberrant Sensory Gating of the Primary Somatosensory Cortex Contributes to the Motor Circuit Dysfunction in Paroxysmal Kinesigenic Dyskinesia.Front Neurol. 2018 Oct 15;9:831. doi: 10.3389/fneur.2018.00831. eCollection 2018. Front Neurol. 2018. PMID: 30386286 Free PMC article.
-
The epileptic and nonepileptic spectrum of paroxysmal dyskinesias: Channelopathies, synaptopathies, and transportopathies.Mov Disord. 2017 Mar;32(3):310-318. doi: 10.1002/mds.26901. Epub 2017 Jan 16. Mov Disord. 2017. PMID: 28090678 Free PMC article. Review.
References
-
- Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, Zhang QJ, Li HF, Murong SX, Xu J, Wang N, Wu ZY. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nature genetics. 2011;43:1252–1255. - PubMed
-
- Cloarec R, Bruneau N, Rudolf G, Massacrier A, Salmi M, Bataillard M, Boulay C, Caraballo R, Fejerman N, Genton P, Hirsch E, Hunter A, Lesca G, Motte J, Roubertie A, Sanlaville D, et al. PRRT2 links infantile convulsions and paroxysmal dyskinesia with migraine. Neurology. 2012;79:2097–2103. - PMC - PubMed
-
- Dale RC, Gardiner A, Antony J, Houlden H. Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine. Dev Med Child Neurol. 2012;54:958–960. - PubMed
-
- Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, Wali GM, Counihan T, Schapira AH, Spacey SD, Valente EM, Silveira-Moriyama L, Teive HA, Raskin S, Sander JW, Lees A, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology. 2012;79:2115–2121. - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
