

The PUF binding landscape in metazoan germ cells
- PMID: 27165521
- PMCID: PMC4911911
- DOI: 10.1261/rna.055871.116
The PUF binding landscape in metazoan germ cells
Abstract
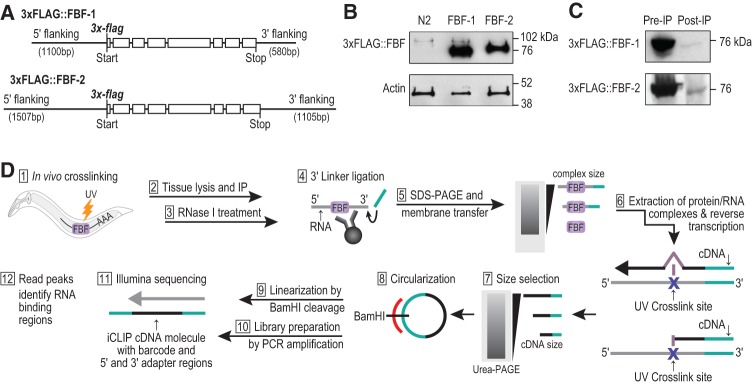
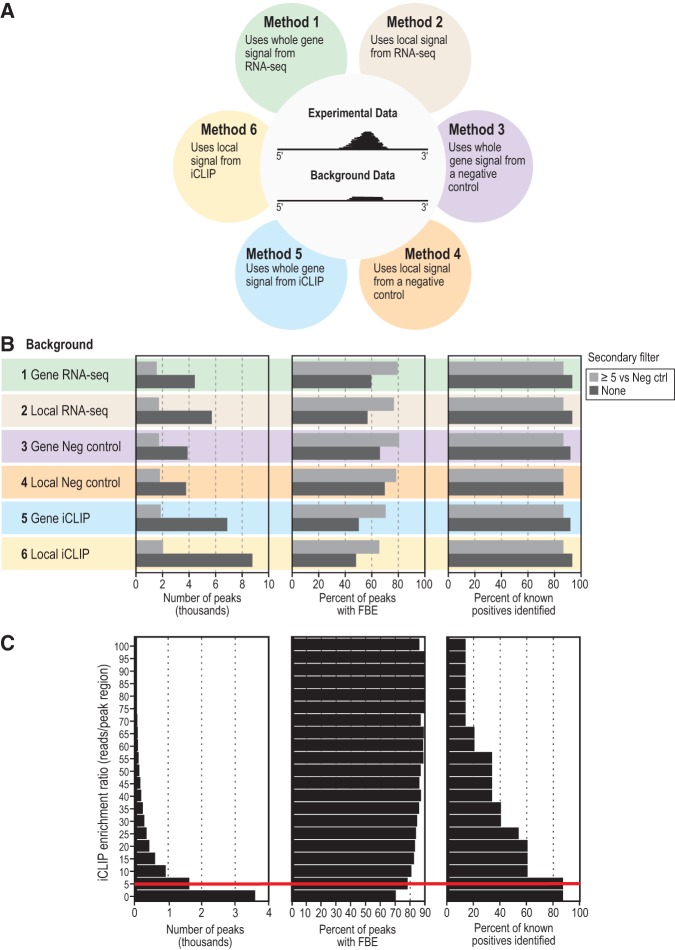
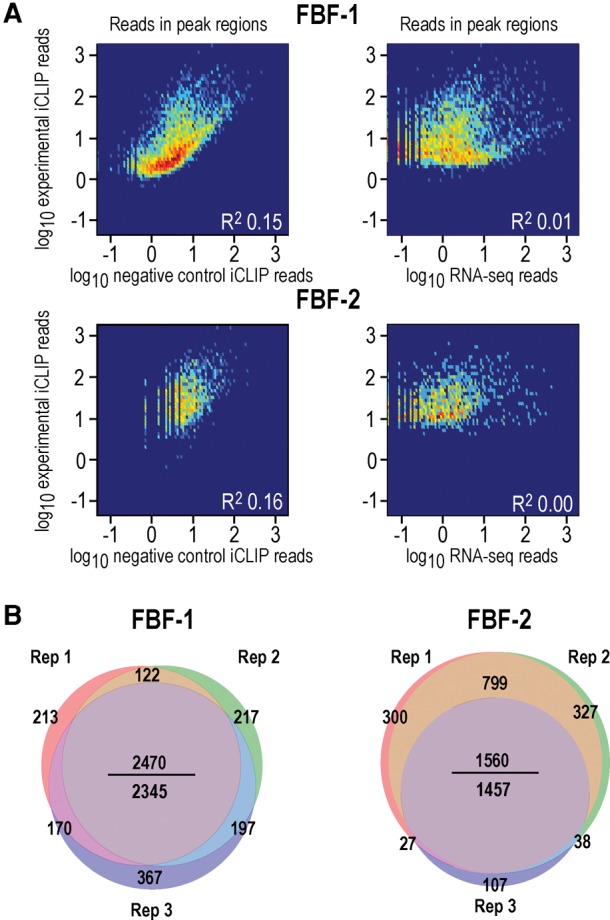
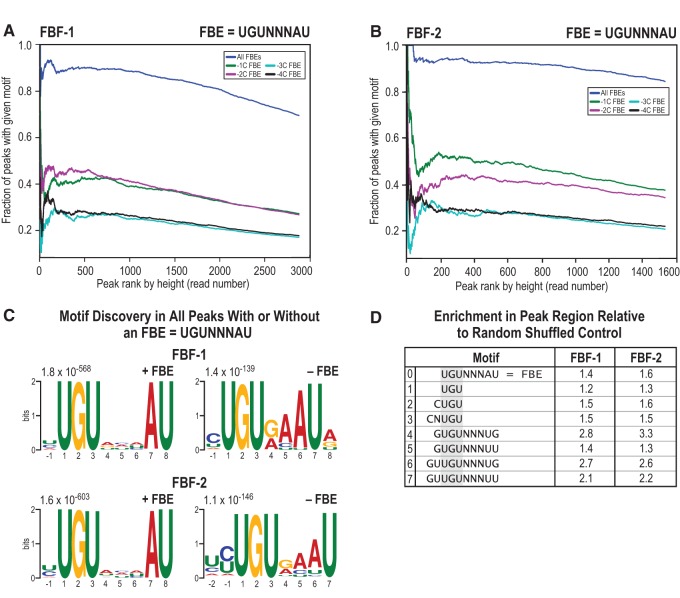
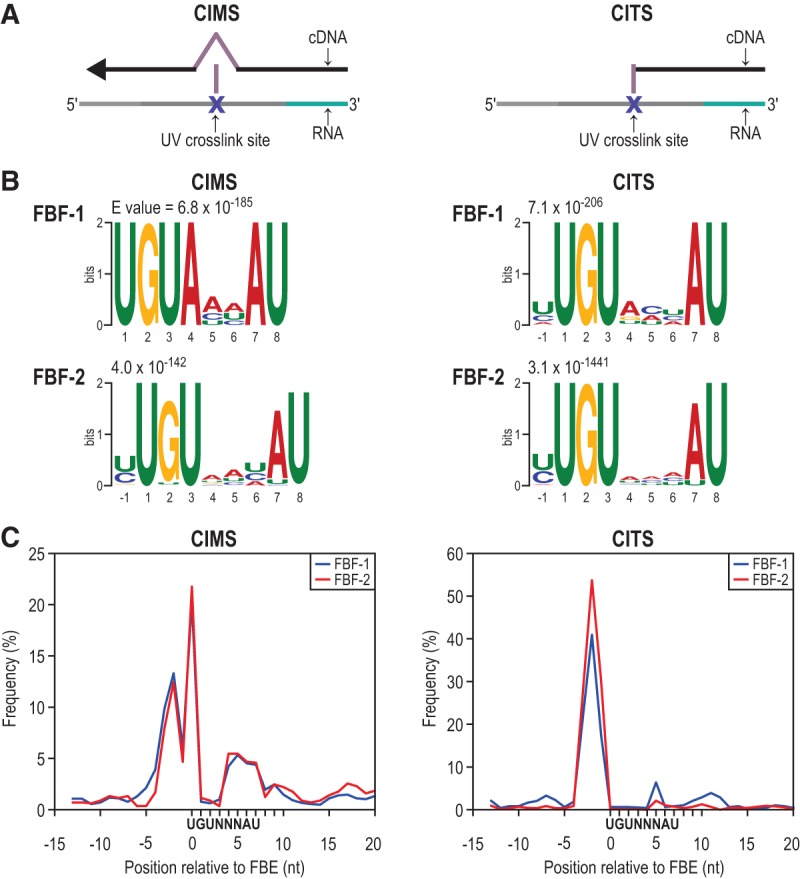
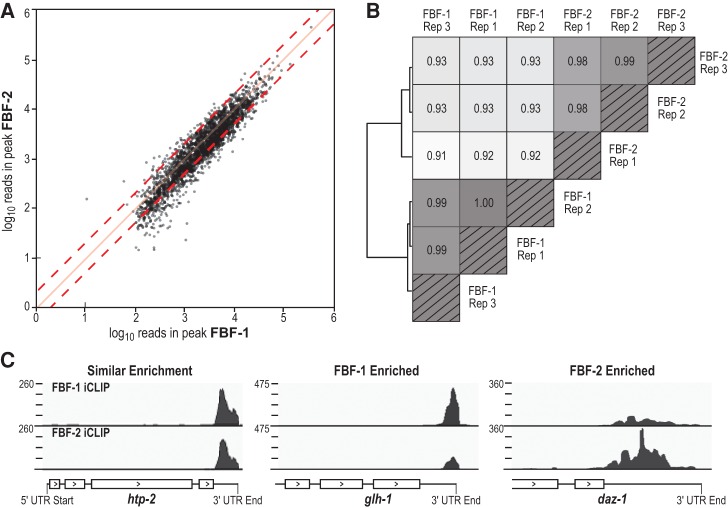
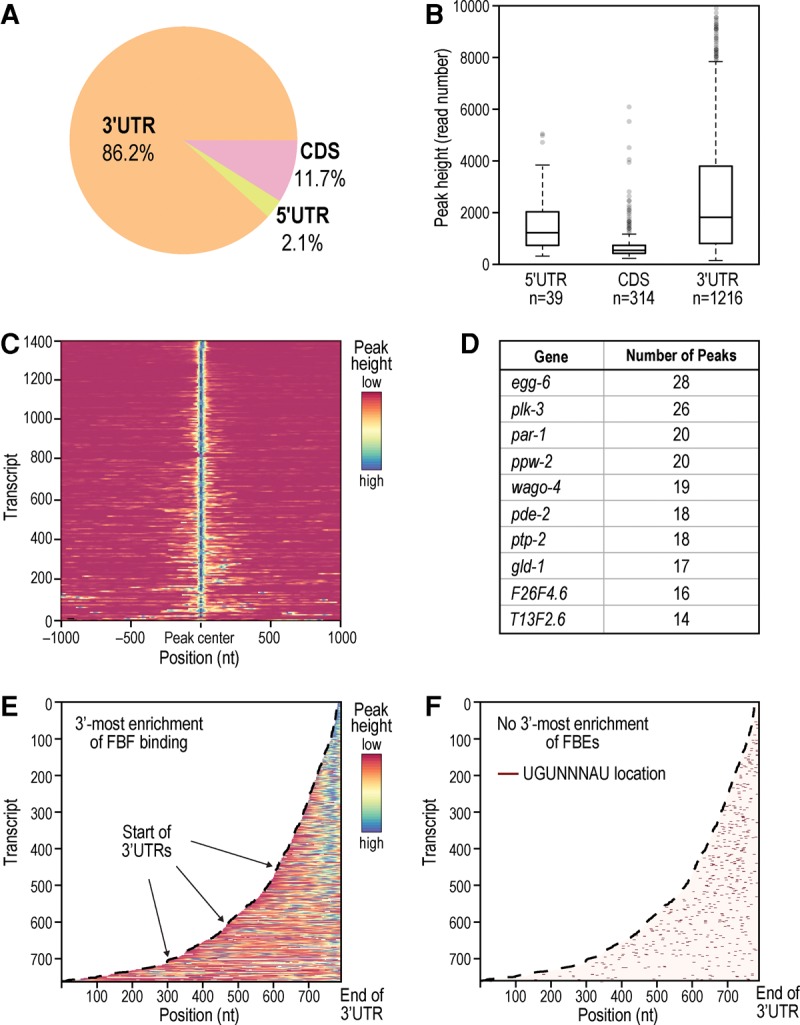
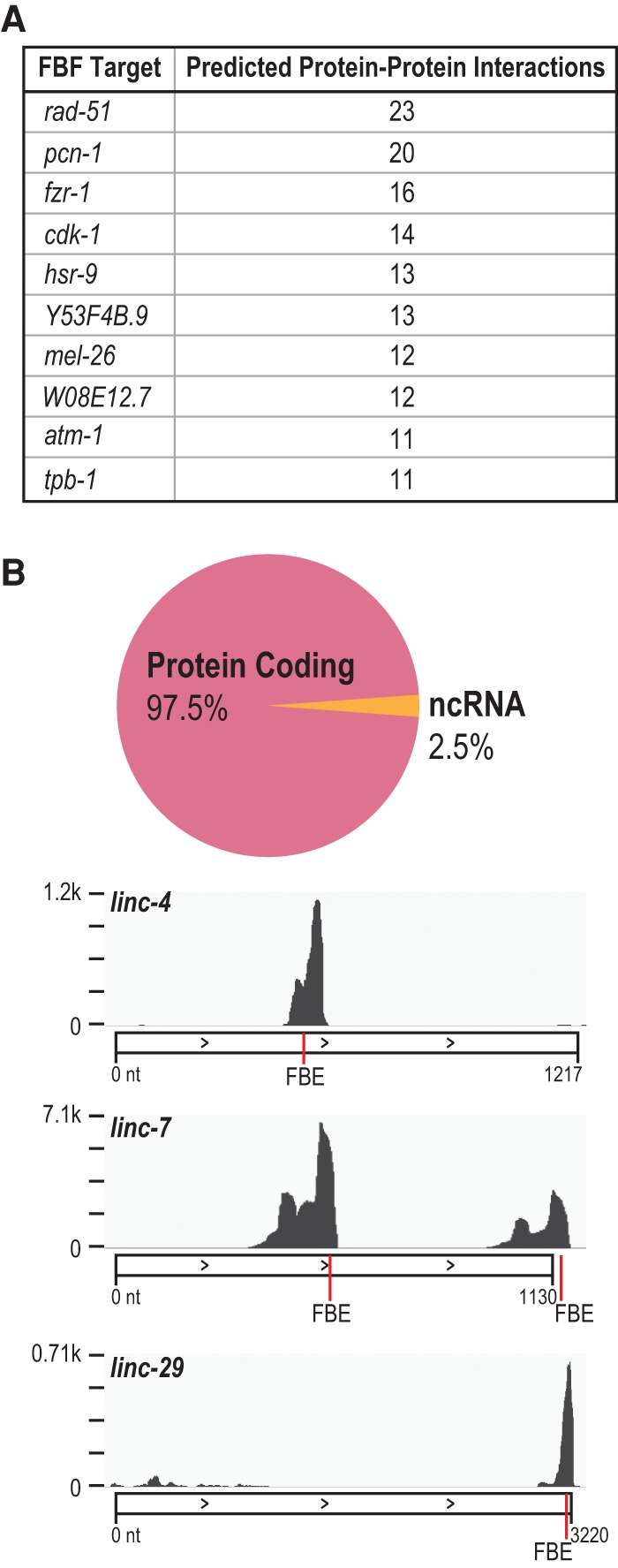
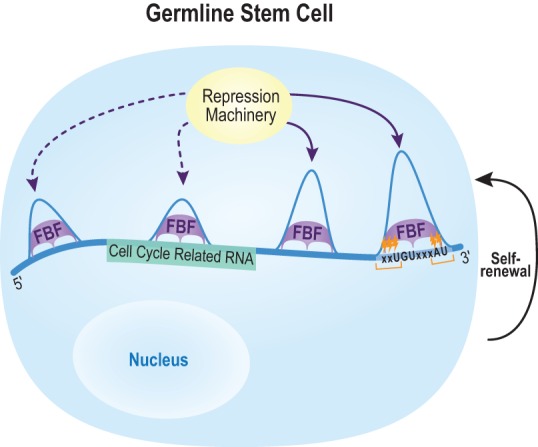
PUF (Pumilio/FBF) proteins are RNA-binding proteins and conserved stem cell regulators. The Caenorhabditis elegans PUF proteins FBF-1 and FBF-2 (collectively FBF) regulate mRNAs in germ cells. Without FBF, adult germlines lose all stem cells. A major gap in our understanding of PUF proteins, including FBF, is a global view of their binding sites in their native context (i.e., their "binding landscape"). To understand the interactions underlying FBF function, we used iCLIP (individual-nucleotide resolution UV crosslinking and immunoprecipitation) to determine binding landscapes of C. elegans FBF-1 and FBF-2 in the germline tissue of intact animals. Multiple iCLIP peak-calling methods were compared to maximize identification of both established FBF binding sites and positive control target mRNAs in our iCLIP data. We discovered that FBF-1 and FBF-2 bind to RNAs through canonical as well as alternate motifs. We also analyzed crosslinking-induced mutations to map binding sites precisely and to identify key nucleotides that may be critical for FBF-RNA interactions. FBF-1 and FBF-2 can bind sites in the 5'UTR, coding region, or 3'UTR, but have a strong bias for the 3' end of transcripts. FBF-1 and FBF-2 have strongly overlapping target profiles, including mRNAs and noncoding RNAs. From a statistically robust list of 1404 common FBF targets, 847 were previously unknown, 154 were related to cell cycle regulation, three were lincRNAs, and 335 were shared with the human PUF protein PUM2.
Keywords: C. elegans; FBF; PUF proteins; RNA; RNA-binding proteins; iCLIP; peak-calling methods; target mRNAs.
© 2016 Prasad et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
Similar articles
-
Binding specificity and mRNA targets of a C. elegans PUF protein, FBF-1.RNA. 2005 Apr;11(4):447-58. doi: 10.1261/rna.7255805. RNA. 2005. PMID: 15769874 Free PMC article.
-
Germline stem cells and their regulation in the nematode Caenorhabditis elegans.Adv Exp Med Biol. 2013;786:29-46. doi: 10.1007/978-94-007-6621-1_3. Adv Exp Med Biol. 2013. PMID: 23696350
-
Toward Identifying Subnetworks from FBF Binding Landscapes in Caenorhabditis Spermatogenic or Oogenic Germlines.G3 (Bethesda). 2019 Jan 9;9(1):153-165. doi: 10.1534/g3.118.200300. G3 (Bethesda). 2019. PMID: 30459181 Free PMC article.
-
Role of PUF-8/PUF protein in stem cell control, sperm-oocyte decision and cell fate reprogramming.J Cell Physiol. 2014 Oct;229(10):1306-11. doi: 10.1002/jcp.24618. J Cell Physiol. 2014. PMID: 24638209 Review.
-
The PUF Protein Family: Overview on PUF RNA Targets, Biological Functions, and Post Transcriptional Regulation.Int J Mol Sci. 2018 Jan 30;19(2):410. doi: 10.3390/ijms19020410. Int J Mol Sci. 2018. PMID: 29385744 Free PMC article. Review.
Cited by
-
PUF partner interactions at a conserved interface shape the RNA-binding landscape and cell fate in Caenorhabditis elegans.Dev Cell. 2024 Mar 11;59(5):661-675.e7. doi: 10.1016/j.devcel.2024.01.005. Epub 2024 Jan 29. Dev Cell. 2024. PMID: 38290520 Free PMC article.
-
A crystal structure of a collaborative RNA regulatory complex reveals mechanisms to refine target specificity.Elife. 2019 Aug 9;8:e48968. doi: 10.7554/eLife.48968. Elife. 2019. PMID: 31397673 Free PMC article.
-
The molecular basis of LST-1 self-renewal activity and its control of stem cell pool size.Development. 2019 Oct 17;146(20):dev181644. doi: 10.1242/dev.181644. Development. 2019. PMID: 31515205 Free PMC article.
-
Identification of proteins and miRNAs that specifically bind an mRNA in vivo.Nat Commun. 2019 Sep 16;10(1):4205. doi: 10.1038/s41467-019-12050-7. Nat Commun. 2019. PMID: 31527589 Free PMC article.
-
Methods to study RNA-protein interactions.Nat Methods. 2019 Mar;16(3):225-234. doi: 10.1038/s41592-019-0330-1. Epub 2019 Feb 25. Nat Methods. 2019. PMID: 30804549 Free PMC article. Review.
References
-
- Ahringer J, Kimble J. 1991. Control of the sperm-oocyte switch in Caenorhabditis elegans hermaphrodites by the fem-3 3′ untranslated region. Nature 349: 346–348. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous