

Satellite Cells in Muscular Dystrophy - Lost in Polarity
- PMID: 27161598
- PMCID: PMC4885782
- DOI: 10.1016/j.molmed.2016.04.002
Satellite Cells in Muscular Dystrophy - Lost in Polarity
Abstract
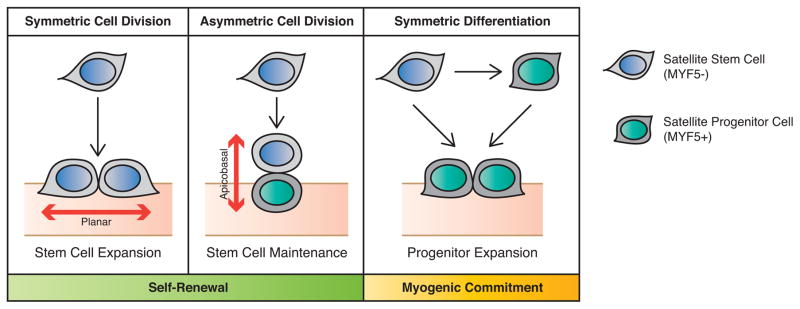
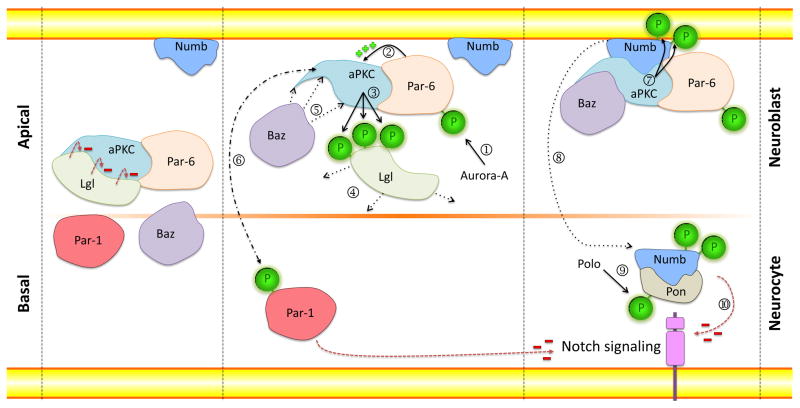
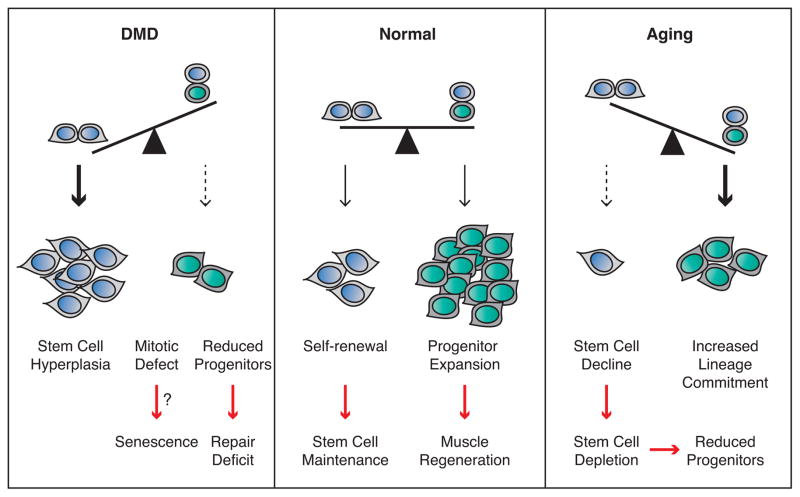
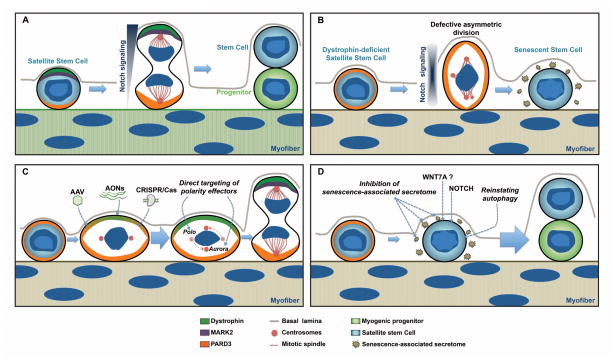
Recent findings employing the mdx mouse model for Duchenne muscular dystrophy (DMD) have revealed that muscle satellite stem cells play a direct role in contributing to disease etiology and progression of DMD, the most common and severe form of muscular dystrophy. Lack of dystrophin expression in DMD has critical consequences in satellite cells including an inability to establish cell polarity, abrogation of asymmetric satellite stem-cell divisions, and failure to enter the myogenic program. Thus, muscle wasting in dystrophic mice is not only caused by myofiber fragility but is exacerbated by intrinsic satellite cell dysfunction leading to impaired regeneration. Despite intense research and clinical efforts, there is still no effective cure for DMD. In this review we highlight recent research advances in DMD and discuss the current state of treatment and, importantly, how we can incorporate satellite cell-targeted therapeutic strategies to correct satellite cell dysfunction in DMD.
Keywords: Duchenne muscular dystrophy; Mark2; Par1b; Pard3; asymmetric division; dystrophin; regenerative myogenesis; satellite cells; stem cell polarity.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division.Nat Med. 2015 Dec;21(12):1455-63. doi: 10.1038/nm.3990. Epub 2015 Nov 16. Nat Med. 2015. PMID: 26569381 Free PMC article.
-
MyD88 is required for satellite cell-mediated myofiber regeneration in dystrophin-deficient mdx mice.Hum Mol Genet. 2018 Oct 1;27(19):3449-3463. doi: 10.1093/hmg/ddy258. Hum Mol Genet. 2018. PMID: 30010933 Free PMC article.
-
CRISPR/Cas9-Based Dystrophin Restoration Reveals a Novel Role for Dystrophin in Bioenergetics and Stress Resistance of Muscle Progenitors.Stem Cells. 2019 Dec;37(12):1615-1628. doi: 10.1002/stem.3094. Epub 2019 Nov 18. Stem Cells. 2019. PMID: 31574188 Free PMC article.
-
Empowering Muscle Stem Cells for the Treatment of Duchenne Muscular Dystrophy.Cells Tissues Organs. 2022;211(6):641-654. doi: 10.1159/000514305. Epub 2021 Apr 28. Cells Tissues Organs. 2022. PMID: 33910206 Review.
-
Epigenetic modifications in muscle regeneration and progression of Duchenne muscular dystrophy.Clin Epigenetics. 2021 Jan 19;13(1):13. doi: 10.1186/s13148-021-01001-z. Clin Epigenetics. 2021. PMID: 33468200 Free PMC article. Review.
Cited by
-
Autophagy and Stem Cells: Self-Eating for Self-Renewal.Front Cell Dev Biol. 2020 Mar 4;8:138. doi: 10.3389/fcell.2020.00138. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32195258 Free PMC article. Review.
-
Retinoic acid maintains human skeletal muscle progenitor cells in an immature state.Cell Mol Life Sci. 2017 May;74(10):1923-1936. doi: 10.1007/s00018-016-2445-1. Epub 2016 Dec 26. Cell Mol Life Sci. 2017. PMID: 28025671 Free PMC article.
-
Plasticity of the Muscle Stem Cell Microenvironment.Adv Exp Med Biol. 2017;1041:141-169. doi: 10.1007/978-3-319-69194-7_8. Adv Exp Med Biol. 2017. PMID: 29204832 Free PMC article. Review.
-
miR-106b is a novel target to promote muscle regeneration and restore satellite stem cell function in injured Duchenne dystrophic muscle.Mol Ther Nucleic Acids. 2022 Aug 20;29:769-786. doi: 10.1016/j.omtn.2022.08.025. eCollection 2022 Sep 13. Mol Ther Nucleic Acids. 2022. PMID: 36159592 Free PMC article.
-
Extra-osseous Roles of the RANK-RANKL-OPG Axis with a Focus on Skeletal Muscle.Curr Osteoporos Rep. 2024 Dec;22(6):632-650. doi: 10.1007/s11914-024-00890-2. Epub 2024 Sep 26. Curr Osteoporos Rep. 2024. PMID: 39325366 Free PMC article. Review.
References
-
- Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscular disorders : NMD. 1991;1:19–29. - PubMed
-
- Emery AE. The muscular dystrophies. Lancet (London, England) 2002;359:687–695. - PubMed
-
- Koenig M, et al. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. - PubMed
-
- Koenig M, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
