

repgenHMM: a dynamic programming tool to infer the rules of immune receptor generation from sequence data
- PMID: 27153709
- PMCID: PMC4920122
- DOI: 10.1093/bioinformatics/btw112
repgenHMM: a dynamic programming tool to infer the rules of immune receptor generation from sequence data
Abstract
Motivation: The diversity of the immune repertoire is initially generated by random rearrangements of the receptor gene during early T and B cell development. Rearrangement scenarios are composed of random events-choices of gene templates, base pair deletions and insertions-described by probability distributions. Not all scenarios are equally likely, and the same receptor sequence may be obtained in several different ways. Quantifying the distribution of these rearrangements is an essential baseline for studying the immune system diversity. Inferring the properties of the distributions from receptor sequences is a computationally hard problem, requiring enumerating every possible scenario for every sampled receptor sequence.
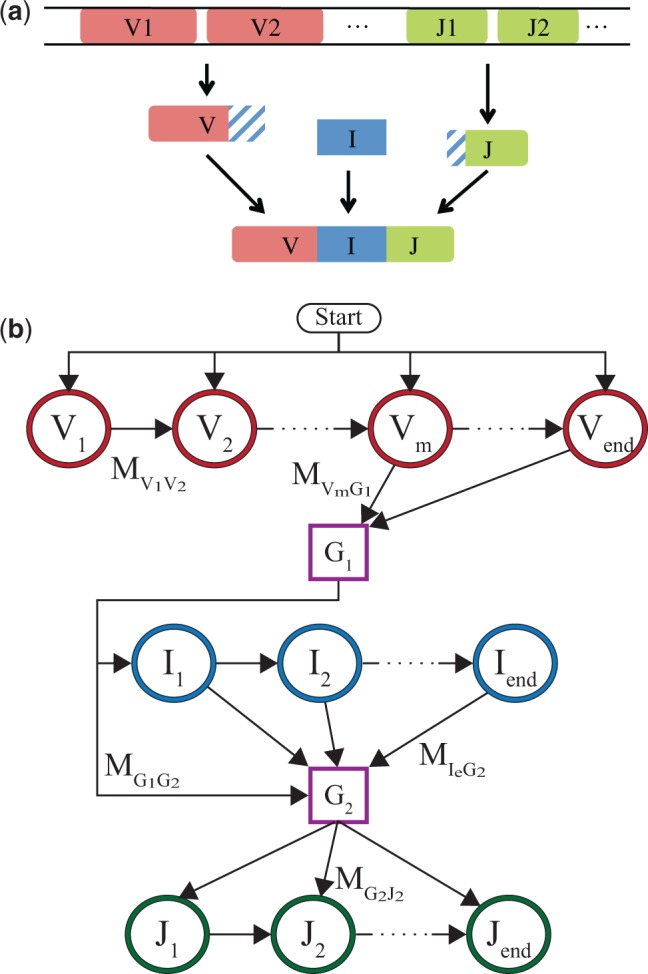
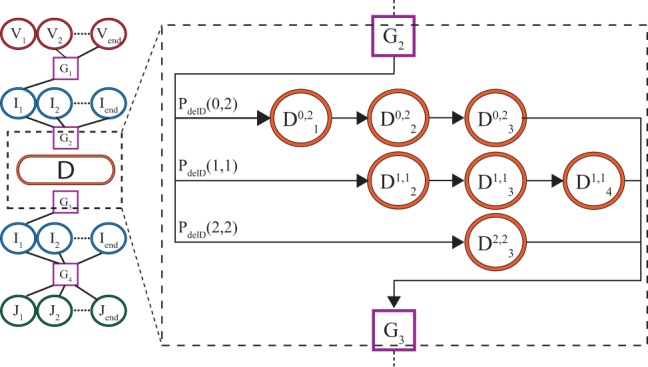
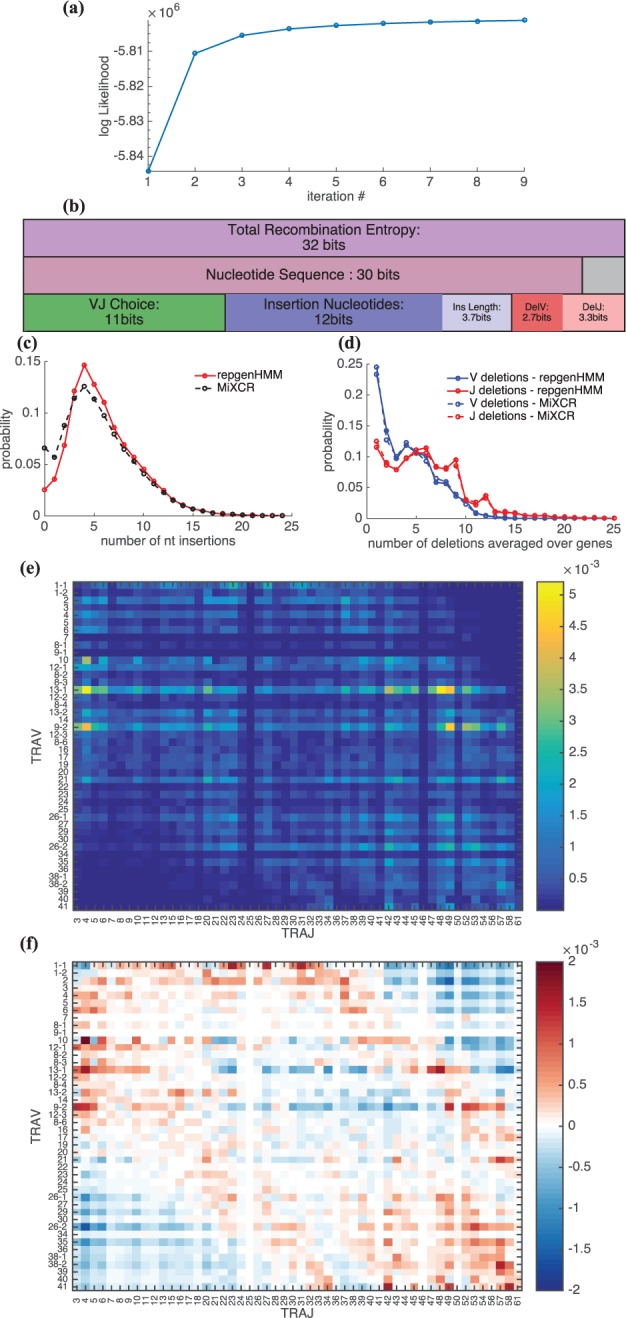
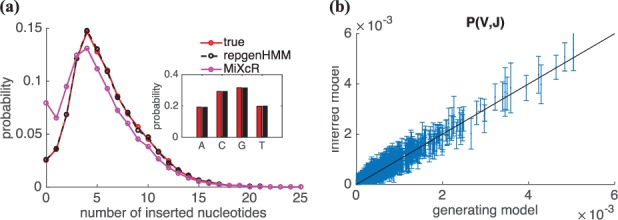
Results: We present a Hidden Markov model, which accounts for all plausible scenarios that can generate the receptor sequences. We developed and implemented a method based on the Baum-Welch algorithm that can efficiently infer the parameters for the different events of the rearrangement process. We tested our software tool on sequence data for both the alpha and beta chains of the T cell receptor. To test the validity of our algorithm, we also generated synthetic sequences produced by a known model, and confirmed that its parameters could be accurately inferred back from the sequences. The inferred model can be used to generate synthetic sequences, to calculate the probability of generation of any receptor sequence, as well as the theoretical diversity of the repertoire. We estimate this diversity to be [Formula: see text] for human T cells. The model gives a baseline to investigate the selection and dynamics of immune repertoires.
Availability and implementation: Source code and sample sequence files are available at https://bitbucket.org/yuvalel/repgenhmm/downloads
Contact: elhanati@lpt.ens.fr or tmora@lps.ens.fr or awalczak@lpt.ens.fr.
© The Author 2016. Published by Oxford University Press.
Figures

Similar articles
-
OLGA: fast computation of generation probabilities of B- and T-cell receptor amino acid sequences and motifs.Bioinformatics. 2019 Sep 1;35(17):2974-2981. doi: 10.1093/bioinformatics/btz035. Bioinformatics. 2019. PMID: 30657870 Free PMC article.
-
Statistical inference of the generation probability of T-cell receptors from sequence repertoires.Proc Natl Acad Sci U S A. 2012 Oct 2;109(40):16161-6. doi: 10.1073/pnas.1212755109. Epub 2012 Sep 17. Proc Natl Acad Sci U S A. 2012. PMID: 22988065 Free PMC article.
-
Recovering probabilities for nucleotide trimming processes for T cell receptor TRA and TRG V-J junctions analyzed with IMGT tools.BMC Bioinformatics. 2008 Oct 2;9:408. doi: 10.1186/1471-2105-9-408. BMC Bioinformatics. 2008. PMID: 18831754 Free PMC article.
-
Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination.Immunol Rev. 2018 Jul;284(1):167-179. doi: 10.1111/imr.12665. Immunol Rev. 2018. PMID: 29944757 Free PMC article. Review.
-
Application of the molecular analysis of the T-cell receptor repertoire in the study of immune-mediated hematologic diseases.Hematology. 2003 Jun;8(3):173-81. doi: 10.1080/1024533031000107505. Hematology. 2003. PMID: 12745651 Review.
Cited by
-
Benchmarking data-driven filtering for denoising of TCRpMHC single-cell data.Sci Rep. 2023 Sep 26;13(1):16147. doi: 10.1038/s41598-023-43048-3. Sci Rep. 2023. PMID: 37752190 Free PMC article.
-
Antibody repertoire sequencing analysis.Acta Biochim Biophys Sin (Shanghai). 2022 May 25;54(6):864-873. doi: 10.3724/abbs.2022062. Acta Biochim Biophys Sin (Shanghai). 2022. PMID: 35713313 Free PMC article. Review.
-
Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires.Front Immunol. 2018 Feb 21;9:224. doi: 10.3389/fimmu.2018.00224. eCollection 2018. Front Immunol. 2018. PMID: 29515569 Free PMC article. Review.
-
Likelihood-Based Inference of B Cell Clonal Families.PLoS Comput Biol. 2016 Oct 17;12(10):e1005086. doi: 10.1371/journal.pcbi.1005086. eCollection 2016 Oct. PLoS Comput Biol. 2016. PMID: 27749910 Free PMC article.
-
OLGA: fast computation of generation probabilities of B- and T-cell receptor amino acid sequences and motifs.Bioinformatics. 2019 Sep 1;35(17):2974-2981. doi: 10.1093/bioinformatics/btz035. Bioinformatics. 2019. PMID: 30657870 Free PMC article.
References
-
- Bishop C.M. (2006). Pattern Recognition and Machine Learning. New York, USA: Springer.
-
- Bolotin D.A. et al. (2012) Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms. Eur. J. Immunol., 42, 3073–3083. - PubMed
-
- Bolotin D.A. et al. (2015) MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods, 12, 380–381. - PubMed
-
- Bonissone S, Pevzner P. (2015). Immunoglobulin classification using the colored antibody graph In: Przytycka T.M. (ed.) Research in Computational Molecular Biology SE - 7, volume 9029 of Lecture Notes in Computer Science. Switzerland: Springer International Publishing, pp. 44–59.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
