

AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells via Activation of CAMKK2
- PMID: 27141100
- PMCID: PMC5390849
- DOI: 10.1158/1541-7786.MCR-15-0479
AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells via Activation of CAMKK2
Abstract
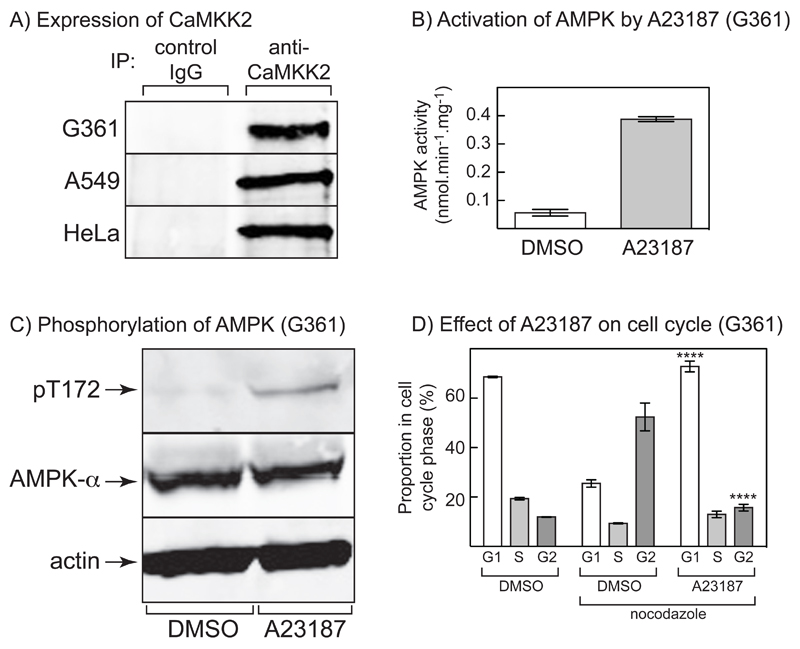
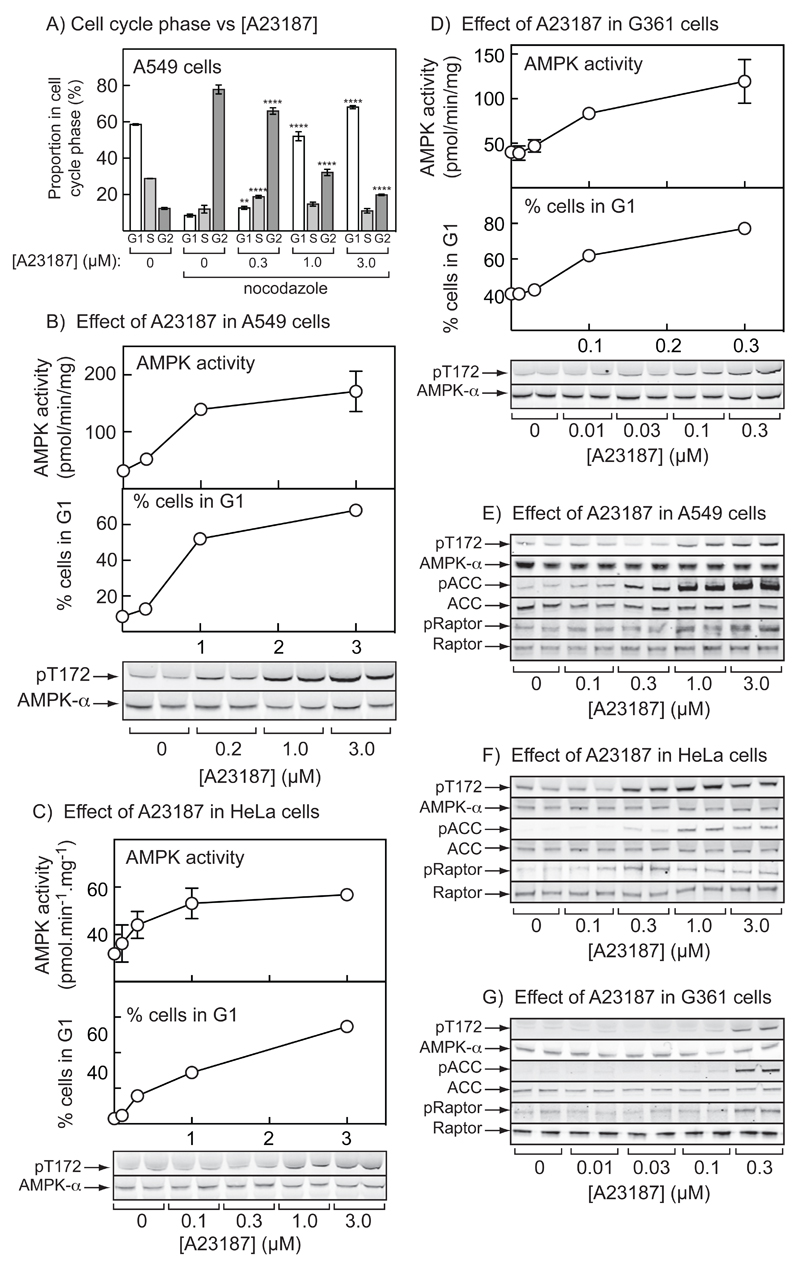
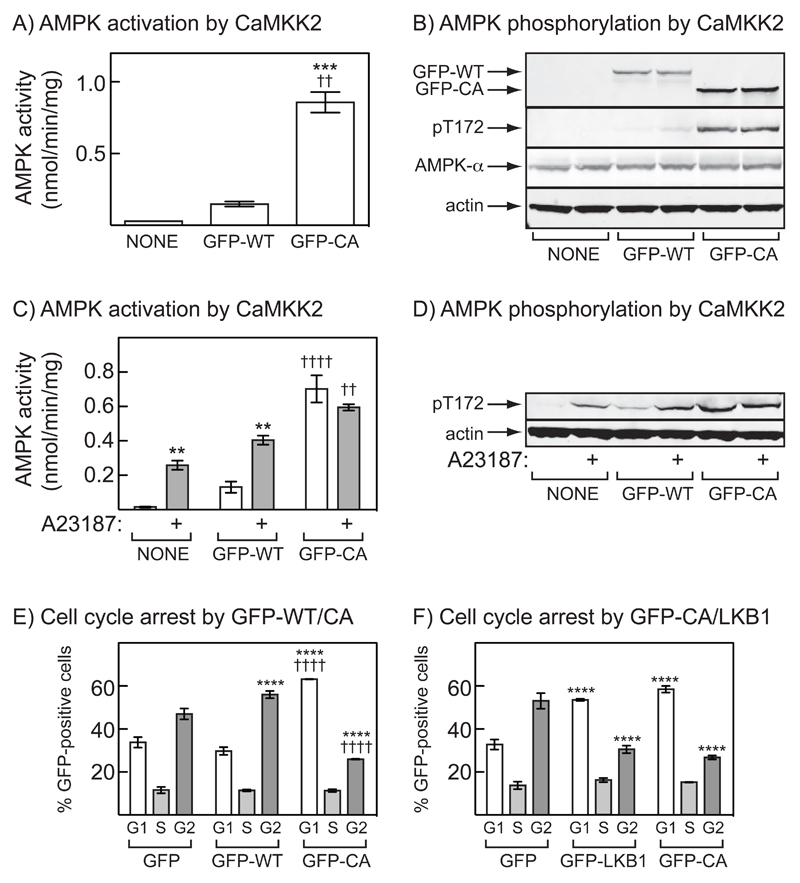
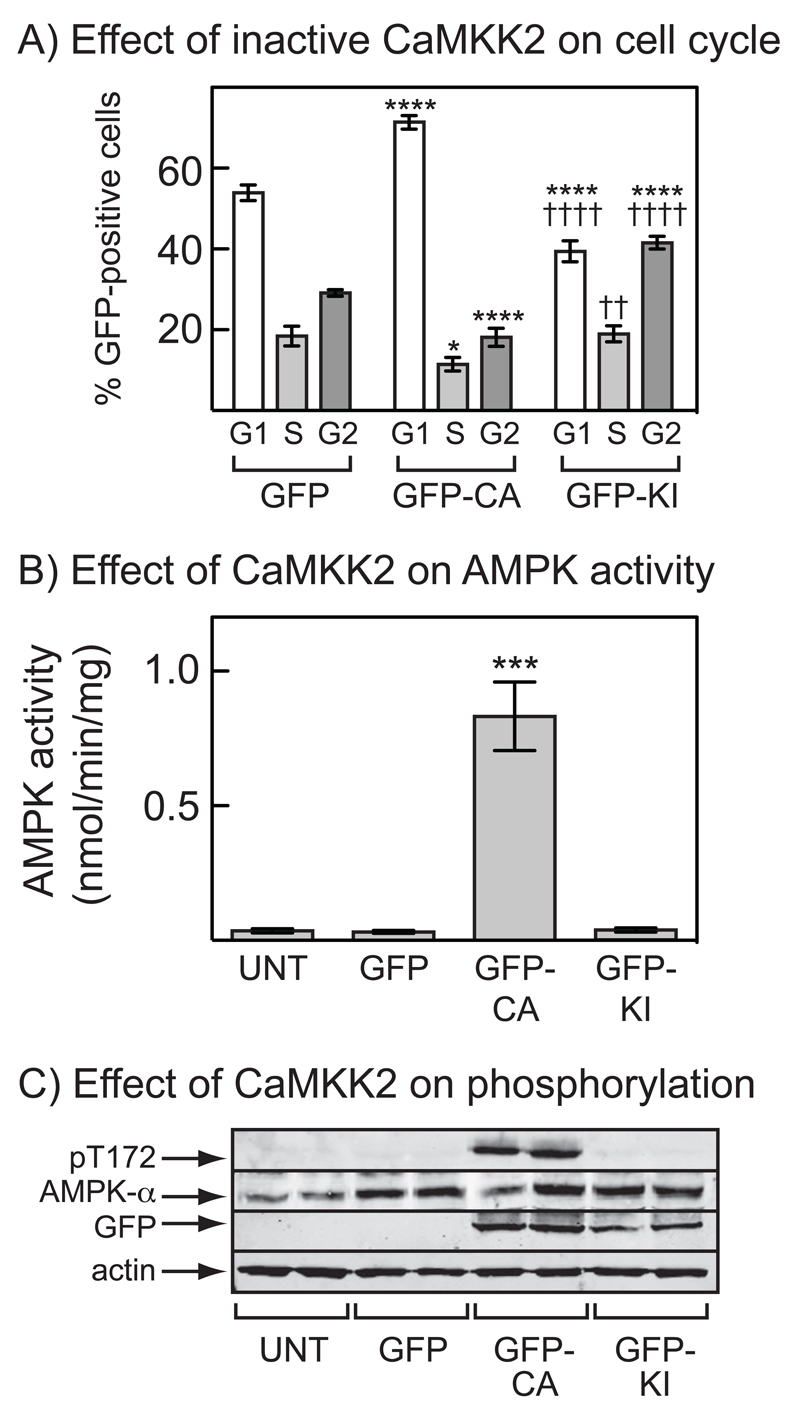
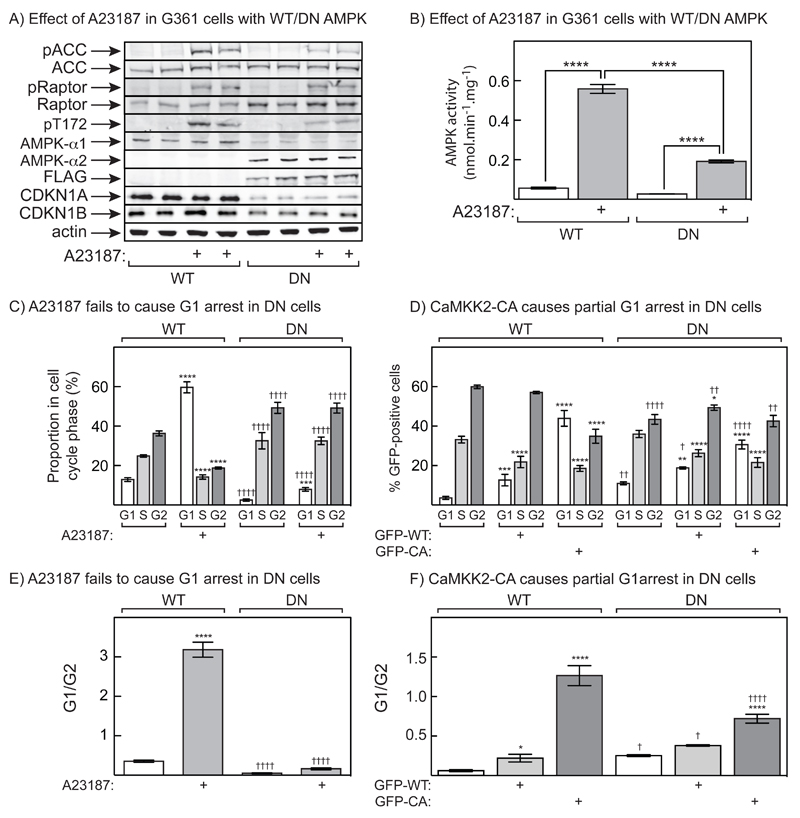
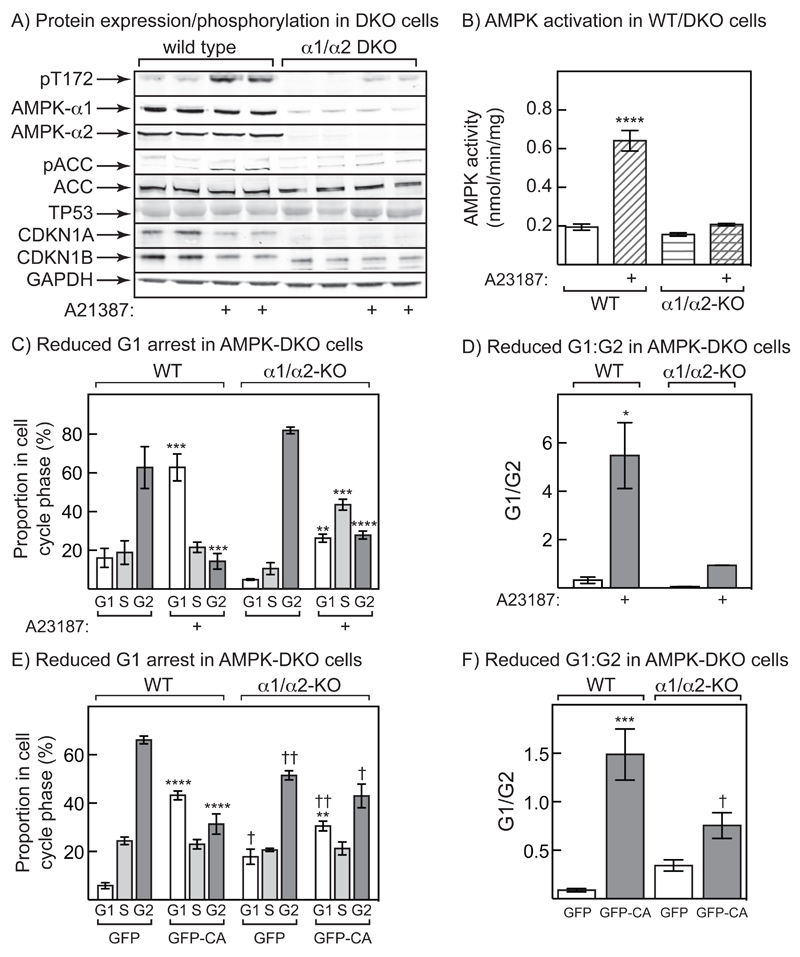
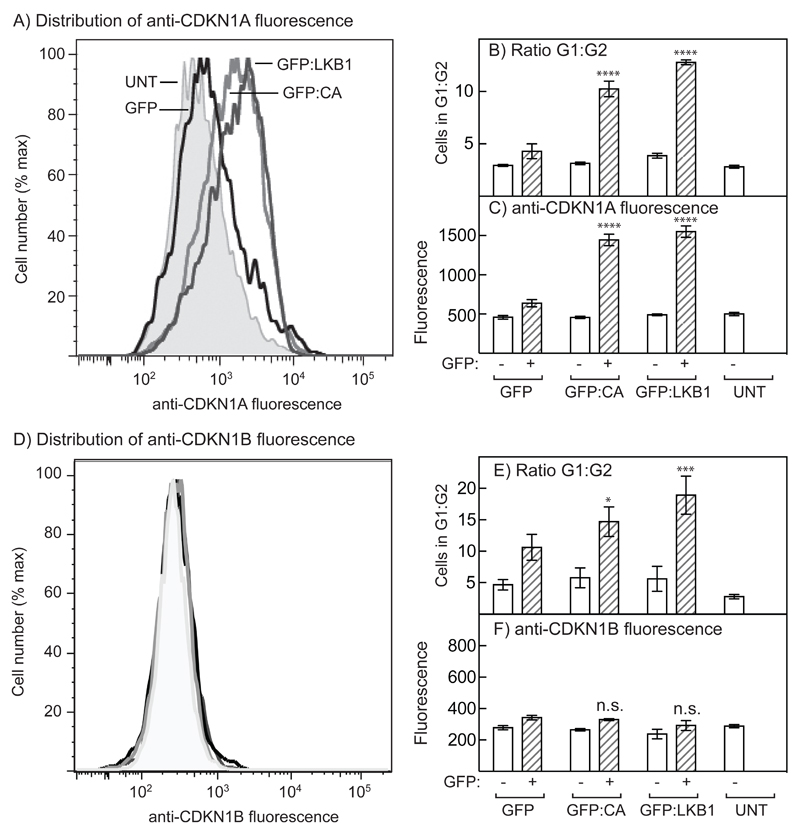
The AMP-activated protein kinase (AMPK) is activated by phosphorylation at Thr172, either by the tumor suppressor kinase LKB1 or by an alternate pathway involving the Ca(2+)/calmodulin-dependent kinase, CAMKK2. Increases in AMP:ATP and ADP:ATP ratios, signifying energy deficit, promote allosteric activation and net Thr172 phosphorylation mediated by LKB1, so that the LKB1-AMPK pathway acts as an energy sensor. Many tumor cells carry loss-of-function mutations in the STK11 gene encoding LKB1, but LKB1 reexpression in these cells causes cell-cycle arrest. Therefore, it was investigated as to whether arrest by LKB1 is caused by activation of AMPK or of one of the AMPK-related kinases, which are also dependent on LKB1 but are not activated by CAMKK2. In three LKB1-null tumor cell lines, treatment with the Ca(2+) ionophore A23187 caused a G1 arrest that correlated with AMPK activation and Thr172 phosphorylation. In G361 cells, expression of a truncated, Ca(2+)/calmodulin-independent CAMKK2 mutant also caused G1 arrest similar to that caused by expression of LKB1, while expression of a dominant-negative AMPK mutant, or a double knockout of both AMPK-α subunits, also prevented the cell-cycle arrest caused by A23187. These mechanistic findings confirm that AMPK activation triggers cell-cycle arrest, and also suggest that the rapid proliferation of LKB1-null tumor cells is due to lack of the restraining influence of AMPK. However, cell-cycle arrest can be restored by reexpressing LKB1 or a constitutively active CAMKK2, or by pharmacologic agents that increase intracellular Ca(2+) and thus activate endogenous CAMKK2.
Implications: Evidence here reveals that the rapid growth and proliferation of cancer cells lacking the tumor suppressor LKB1 is due to reduced activity of AMPK, and suggests a therapeutic approach by which this block might be circumvented. Mol Cancer Res; 14(8); 683-95. ©2016 AACR.
©2016 American Association for Cancer Research.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
Similar articles
-
Concurrent regulation of LKB1 and CaMKK2 in the activation of AMPK in castrate-resistant prostate cancer by a well-defined polyherbal mixture with anticancer properties.BMC Complement Altern Med. 2018 Jun 18;18(1):188. doi: 10.1186/s12906-018-2255-0. BMC Complement Altern Med. 2018. PMID: 29914450 Free PMC article.
-
CaMKK2 is not involved in contraction-stimulated AMPK activation and glucose uptake in skeletal muscle.Mol Metab. 2023 Sep;75:101761. doi: 10.1016/j.molmet.2023.101761. Epub 2023 Jun 26. Mol Metab. 2023. PMID: 37380024 Free PMC article.
-
Genotoxic Damage Activates the AMPK-α1 Isoform in the Nucleus via Ca2+/CaMKK2 Signaling to Enhance Tumor Cell Survival.Mol Cancer Res. 2018 Feb;16(2):345-357. doi: 10.1158/1541-7786.MCR-17-0323. Epub 2017 Nov 13. Mol Cancer Res. 2018. PMID: 29133590
-
Spatial control of AMPK signaling at subcellular compartments.Crit Rev Biochem Mol Biol. 2020 Feb;55(1):17-32. doi: 10.1080/10409238.2020.1727840. Epub 2020 Feb 18. Crit Rev Biochem Mol Biol. 2020. PMID: 32069425 Free PMC article. Review.
-
AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?Int J Mol Sci. 2020 Dec 27;22(1):186. doi: 10.3390/ijms22010186. Int J Mol Sci. 2020. PMID: 33375416 Free PMC article. Review.
Cited by
-
Embigin Promotes Prostate Cancer Progression by S100A4-Dependent and-Independent Mechanisms.Cancers (Basel). 2018 Jul 23;10(7):239. doi: 10.3390/cancers10070239. Cancers (Basel). 2018. PMID: 30041429 Free PMC article.
-
Oridonin inhibited epithelial-mesenchymal transition of laryngeal carcinoma by positively regulating LKB1/AMPK signaling.Int J Med Sci. 2024 Jan 21;21(4):623-632. doi: 10.7150/ijms.92182. eCollection 2024. Int J Med Sci. 2024. PMID: 38464825 Free PMC article.
-
Sanghuangporus sanghuang Mycelium Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation.Antioxidants (Basel). 2021 Jun 2;10(6):897. doi: 10.3390/antiox10060897. Antioxidants (Basel). 2021. PMID: 34199606 Free PMC article.
-
LKB1 biology: assessing the therapeutic relevancy of LKB1 inhibitors.Cell Commun Signal. 2024 Jun 6;22(1):310. doi: 10.1186/s12964-024-01689-5. Cell Commun Signal. 2024. PMID: 38844908 Free PMC article. Review.
-
Attenuation of Lipopolysaccharide-Induced Acute Lung Injury by Hispolon in Mice, Through Regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 Pathways, and Suppressing Oxidative Stress-Mediated ER Stress-Induced Apoptosis and Autophagy.Nutrients. 2020 Jun 10;12(6):1742. doi: 10.3390/nu12061742. Nutrients. 2020. PMID: 32532087 Free PMC article.
References
-
- Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2014;33C:1–7. - PubMed
-
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, et al. Characterization of the AMP-activated protein kinase kinase from rat liver, and identification of threonine-172 as the major site at which it phosphorylates and activates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–87. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
