

Structure-activity relationship study of angiotensin II analogs in terms of β-arrestin-dependent signaling to aldosterone production
- PMID: 27069636
- PMCID: PMC4804318
- DOI: 10.1002/prp2.226
Structure-activity relationship study of angiotensin II analogs in terms of β-arrestin-dependent signaling to aldosterone production
Abstract
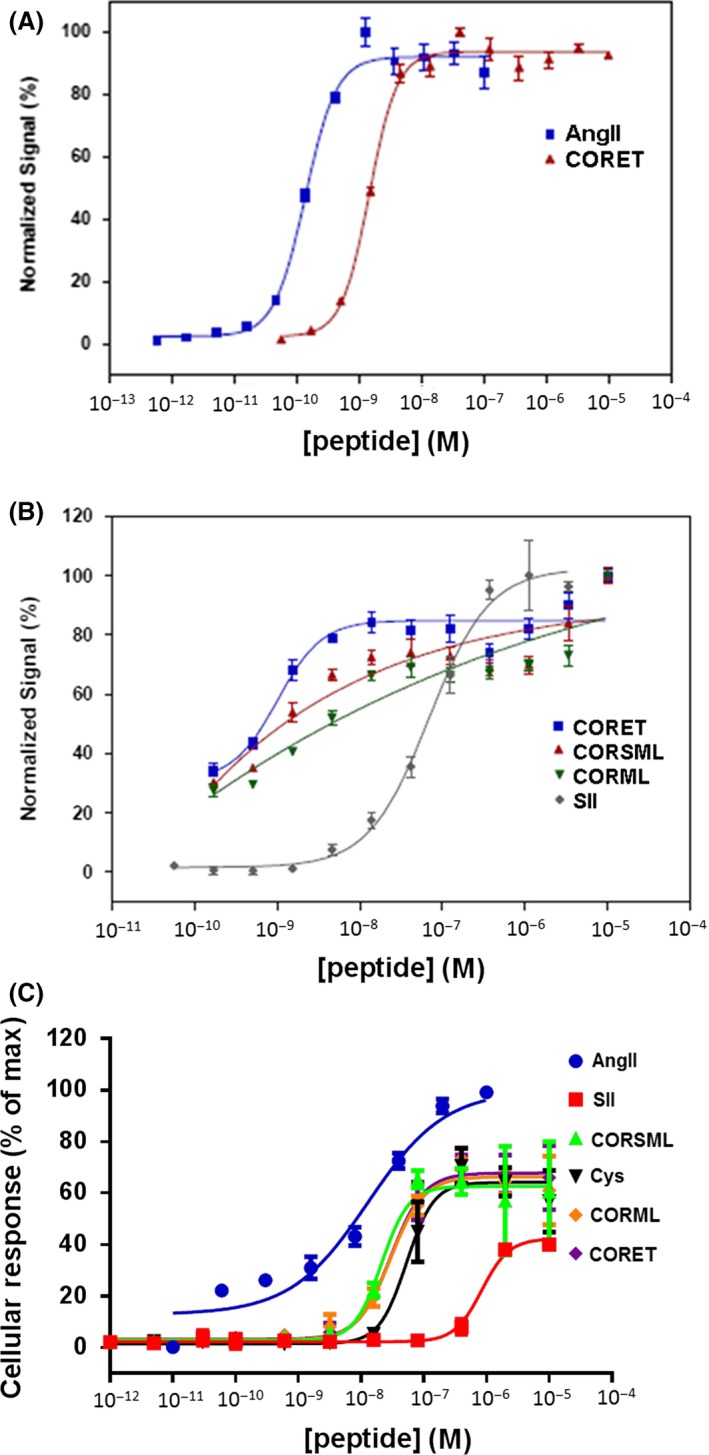
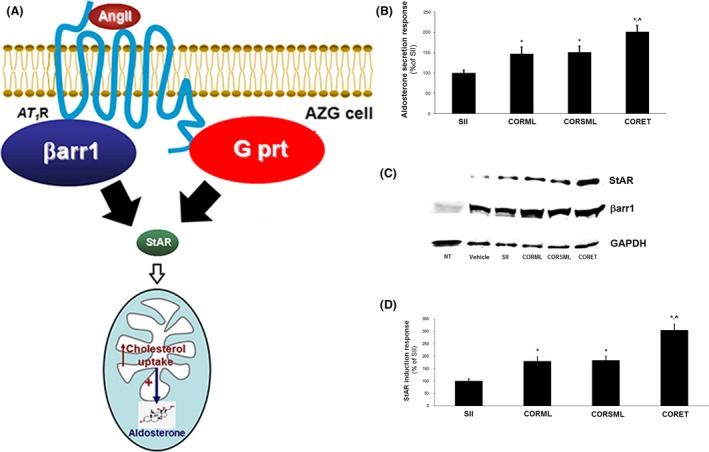
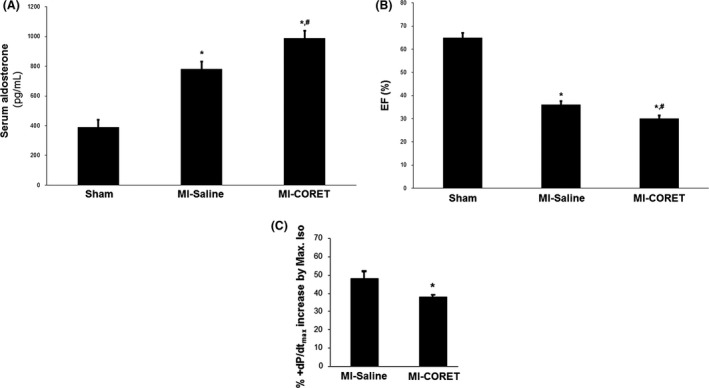
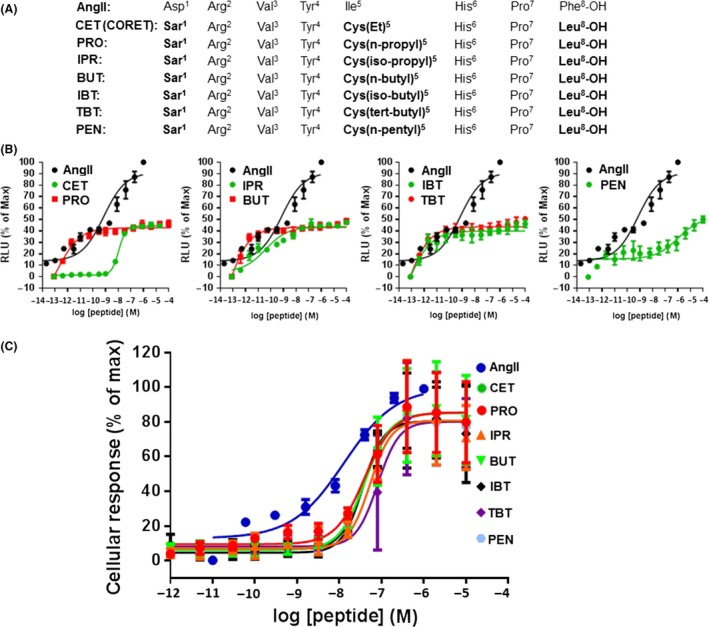
The known angiotensin II (AngII) physiological effect of aldosterone synthesis and secretion induction, a steroid hormone that contributes to the pathology of postmyocardial infarction (MI) heart failure (HF), is mediated by both Gq/11 proteins and β-arrestins, both of which couple to the AngII type 1 receptors (AT1Rs) of adrenocortical zona glomerulosa (AZG) cells. Over the past several years, AngII analogs with increased selectivity ("bias") toward β-arrestin-dependent signaling at the AT1R have been designed and described, starting with SII, the gold-standard β-arrestin-"biased" AngII analog. In this study, we examined the relative potencies of an extensive series of AngII peptide analogs at relative activation of G proteins versus β-arrestins by the AT1R. The major structural difference of these peptides from SII was their varied substitutions at position 5, rather than position 4 of native AngII. Three of them were found biased for β-arrestin activation and extremely potent at stimulating aldosterone secretion in AZG cells in vitro, much more potent than SII in that regard. Finally, the most potent of these three ([Sar(1), Cys(Et)(5), Leu(8)]-AngII, CORET) was further examined in post-MI rats progressing to HF and overexpressing adrenal β-arrestin1 in vivo. Consistent with the in vitro studies, CORET was found to exacerbate the post-MI hyperaldosteronism, and, consequently, cardiac function of the post-MI animals in vivo. Finally, our data suggest that increasing the size of position 5 of the AngII peptide sequence results in directly proportional increases in AT1R-dependent β-arrestin activation. These findings provide important insights for AT1R pharmacology and future AngII-targeted drug development.
Keywords: Aldosterone; angiotensin II; angiotensin II type 1 receptor; biased ligand; structure–activity relationship (SAR); β‐arrestin; post‐MI HF.
Figures
Similar articles
-
An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo.Proc Natl Acad Sci U S A. 2009 Apr 7;106(14):5825-30. doi: 10.1073/pnas.0811706106. Epub 2009 Mar 16. Proc Natl Acad Sci U S A. 2009. PMID: 19289825 Free PMC article.
-
Angiotensin receptor blocker drugs and inhibition of adrenal beta-arrestin-1-dependent aldosterone production: Implications for heart failure therapy.World J Cardiol. 2017 Mar 26;9(3):200-206. doi: 10.4330/wjc.v9.i3.200. World J Cardiol. 2017. PMID: 28400916 Free PMC article.
-
GRK2-Mediated Crosstalk Between β-Adrenergic and Angiotensin II Receptors Enhances Adrenocortical Aldosterone Production In Vitro and In Vivo.Int J Mol Sci. 2020 Jan 16;21(2):574. doi: 10.3390/ijms21020574. Int J Mol Sci. 2020. PMID: 31963151 Free PMC article.
-
Adrenal angiotensin II type 1 receptor biased signaling: The case for "biased" inverse agonism for effective aldosterone suppression.Cell Signal. 2021 Jun;82:109967. doi: 10.1016/j.cellsig.2021.109967. Epub 2021 Feb 25. Cell Signal. 2021. PMID: 33640432 Review.
-
Biased agonism/antagonism at the AngII-AT1 receptor: Implications for adrenal aldosterone production and cardiovascular therapy.Pharmacol Res. 2017 Nov;125(Pt A):14-20. doi: 10.1016/j.phrs.2017.05.009. Epub 2017 May 13. Pharmacol Res. 2017. PMID: 28511989 Review.
Cited by
-
Nicotine-induced adrenal beta-arrestin1 upregulation mediates tobacco-related hyperaldosteronism leading to cardiac dysfunction.World J Cardiol. 2020 May 26;12(5):192-202. doi: 10.4330/wjc.v12.i5.192. World J Cardiol. 2020. PMID: 32547713 Free PMC article.
-
The place of ARBs in heart failure therapy: is aldosterone suppression the key?Ther Adv Cardiovasc Dis. 2019 Jan-Dec;13:1753944719868134. doi: 10.1177/1753944719868134. Ther Adv Cardiovasc Dis. 2019. PMID: 31401939 Free PMC article. Review.
-
Insights Into the Role of Angiotensin-II AT1 Receptor-Dependent β-Arrestin Signaling in Cardiovascular Disease.Hypertension. 2024 Jan;81(1):6-16. doi: 10.1161/HYPERTENSIONAHA.123.19419. Epub 2023 Jul 14. Hypertension. 2024. PMID: 37449411 Free PMC article. Review.
-
GTPγS Assay for Measuring Agonist-Induced Desensitization of Two Human Polymorphic Alpha2B-Adrenoceptor Variants.Methods Mol Biol. 2022;2547:267-273. doi: 10.1007/978-1-0716-2573-6_12. Methods Mol Biol. 2022. PMID: 36068469
-
Not all arrestins are created equal: Therapeutic implications of the functional diversity of the β-arrestins in the heart.World J Cardiol. 2019 Feb 26;11(2):47-56. doi: 10.4330/wjc.v11.i2.47. World J Cardiol. 2019. PMID: 30820275 Free PMC article.
References
-
- Balakumar P, Jagadeesh G (2014). Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol 53: R71–R92. - PubMed
-
- Bomback AS, Klemmer PJ (2007). The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol 3: 486–492. - PubMed
-
- De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52: 415–472. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
