

Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation
- PMID: 27044110
- PMCID: PMC4843475
- DOI: 10.1073/pnas.1602244113
Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation
Abstract
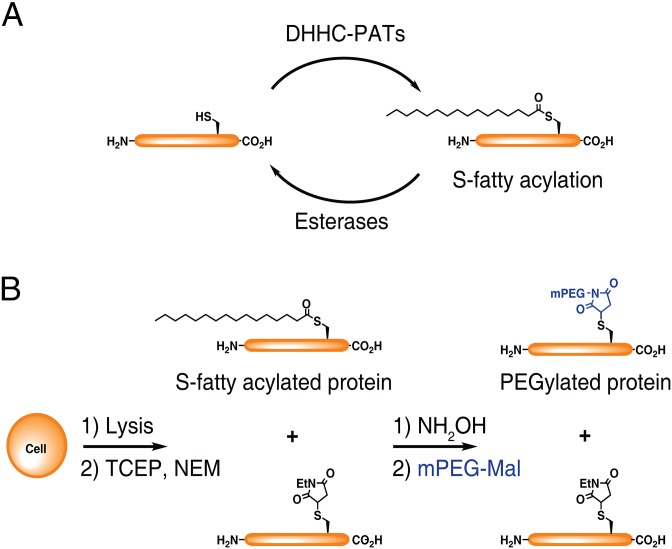
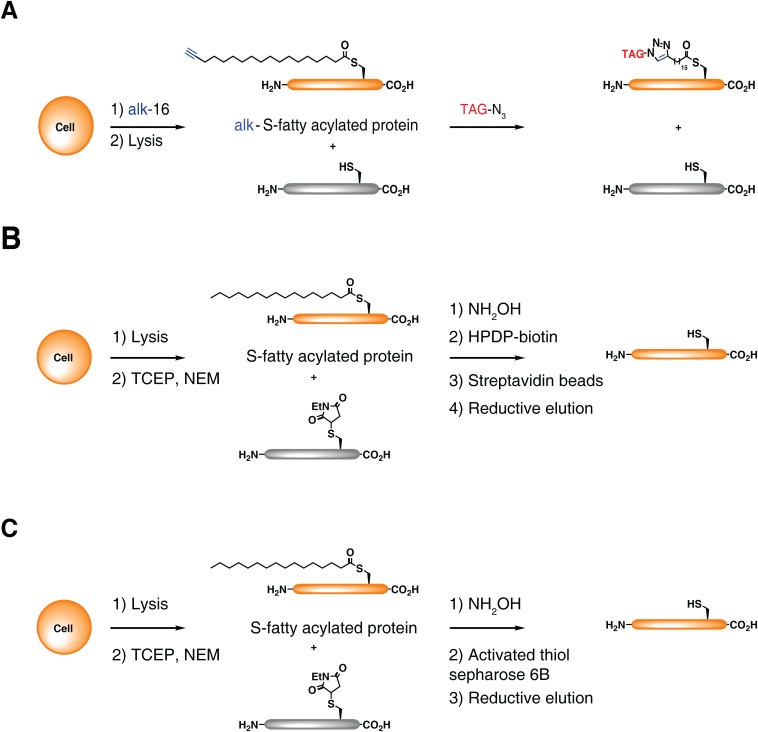
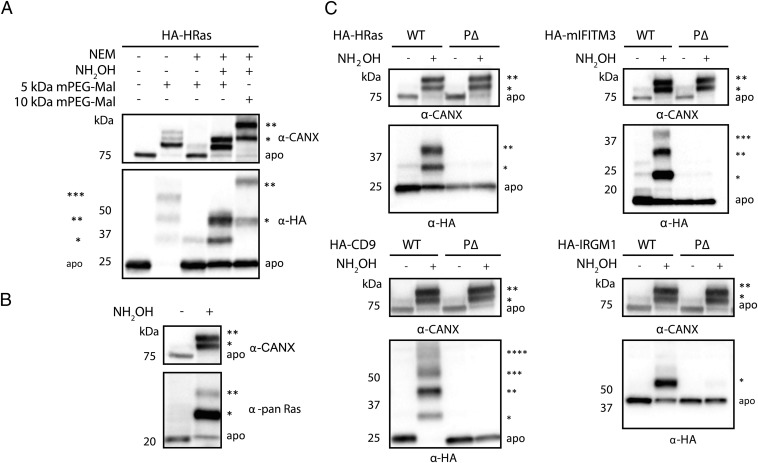
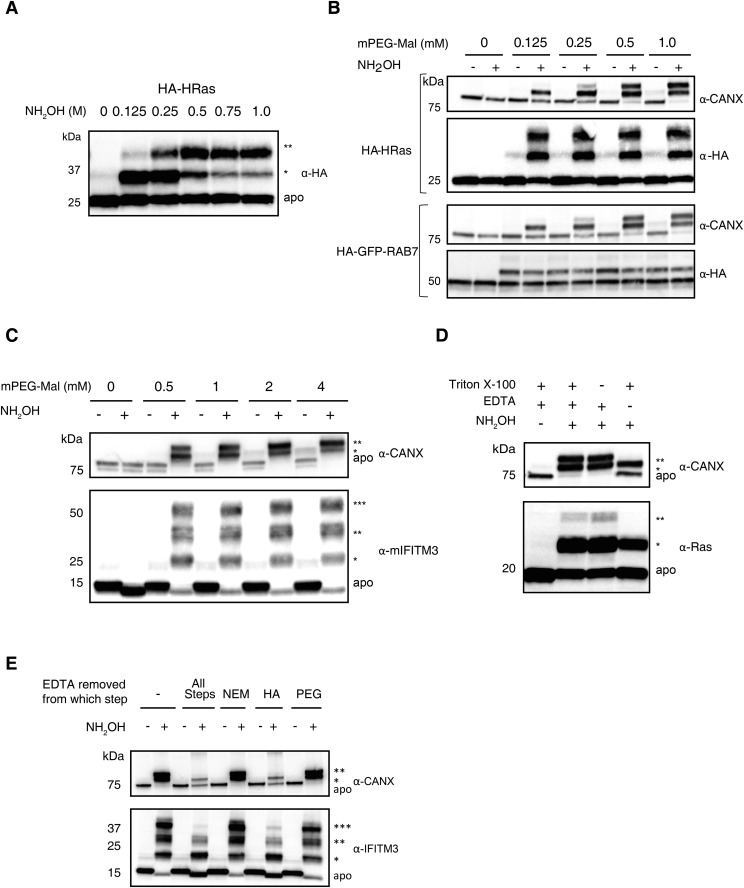
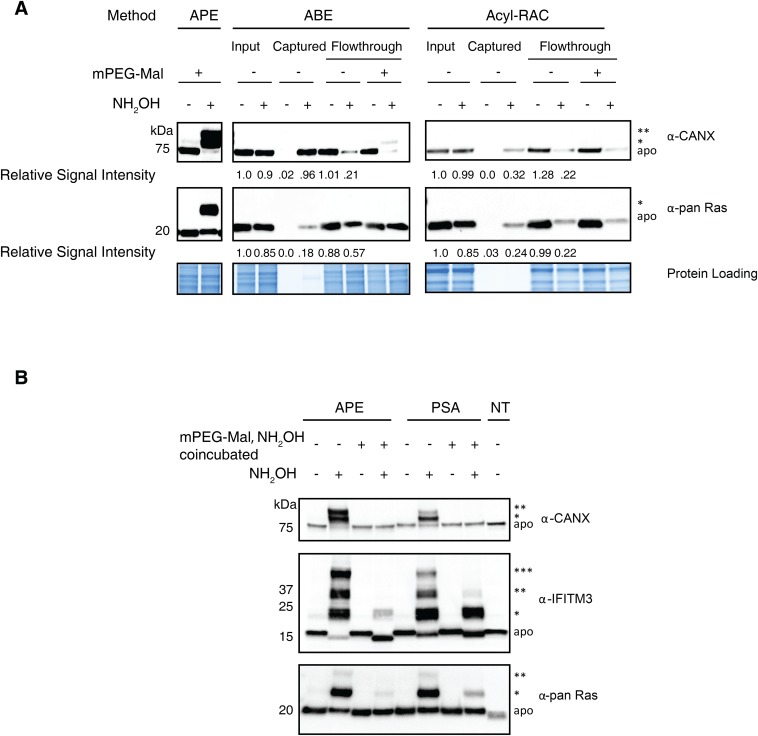
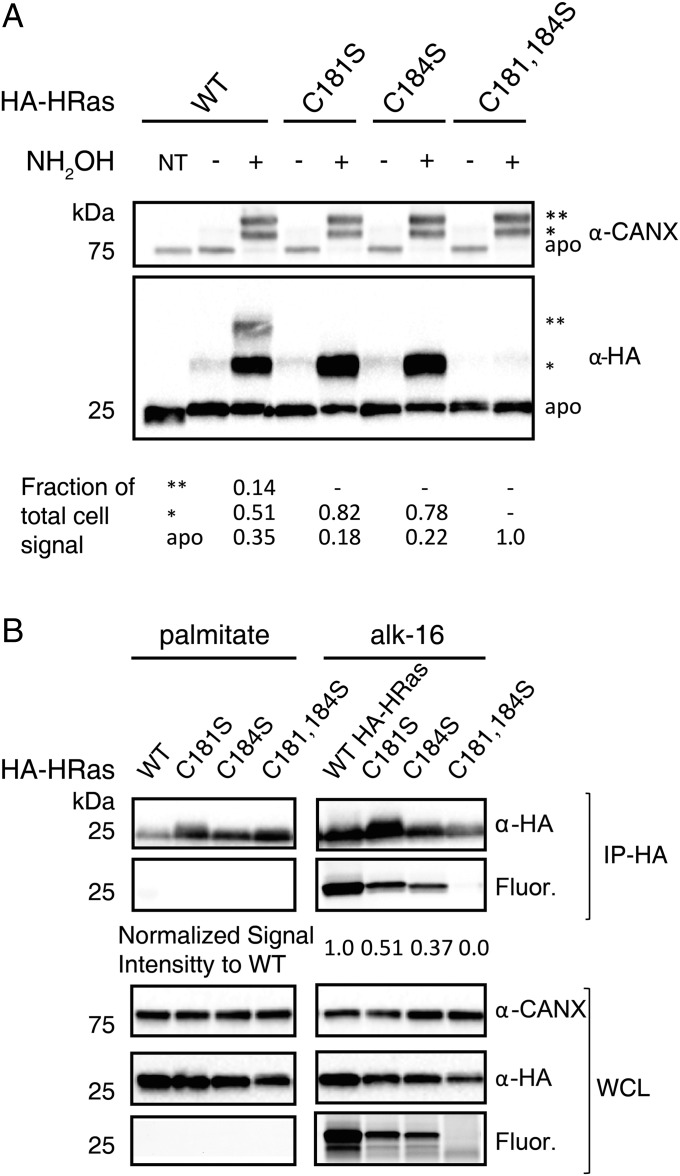
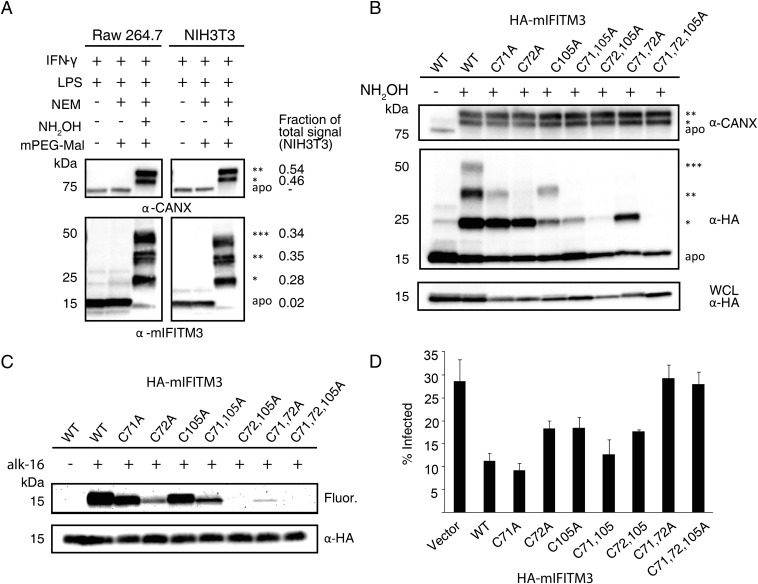
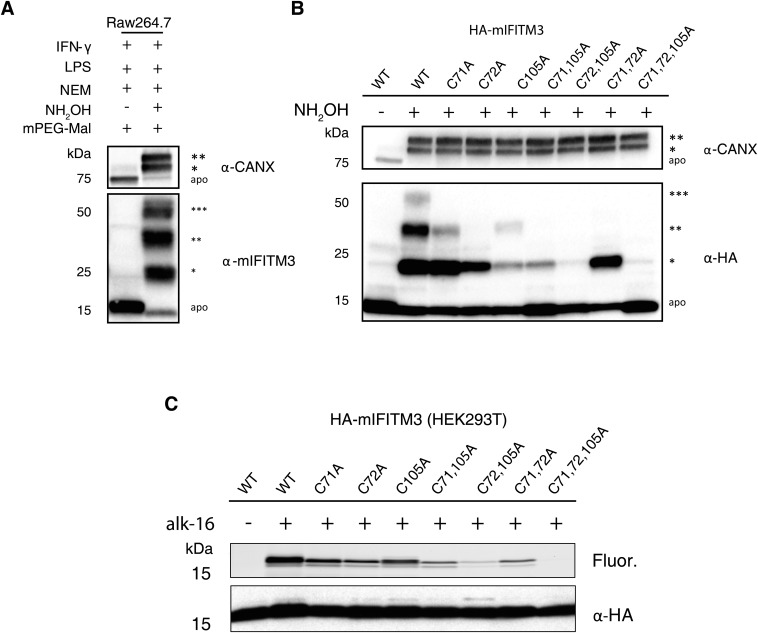
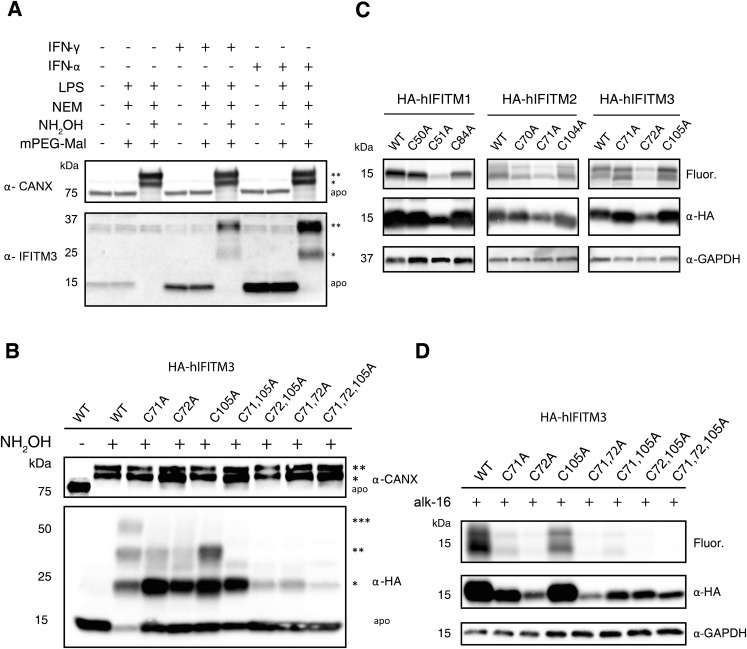
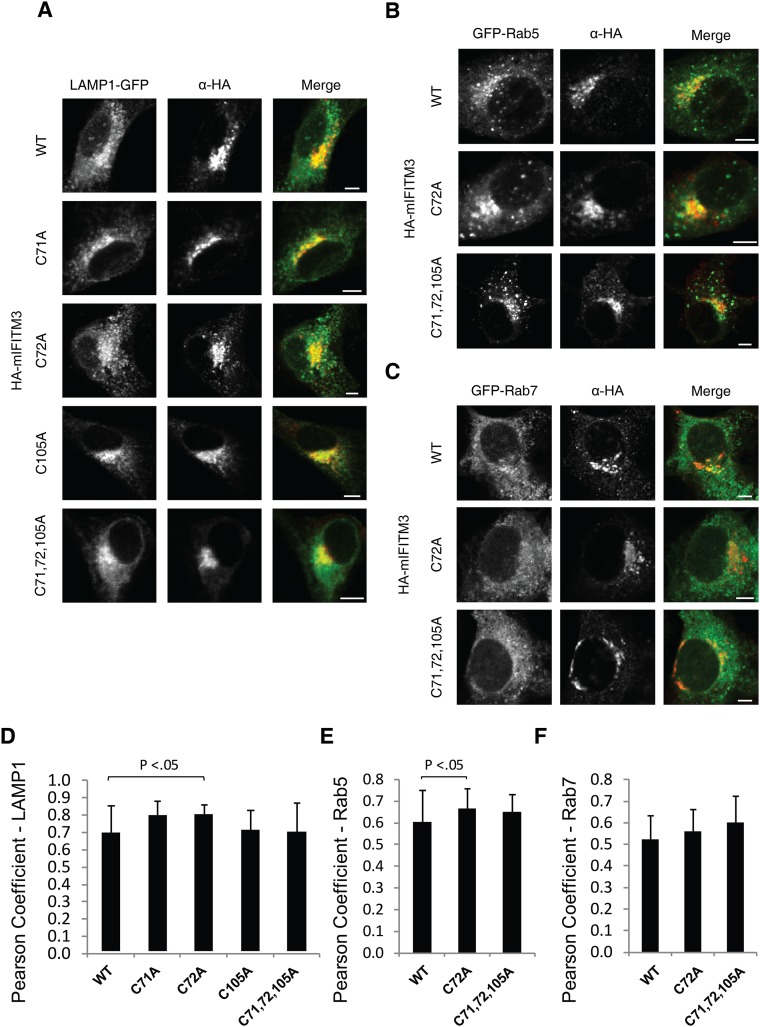
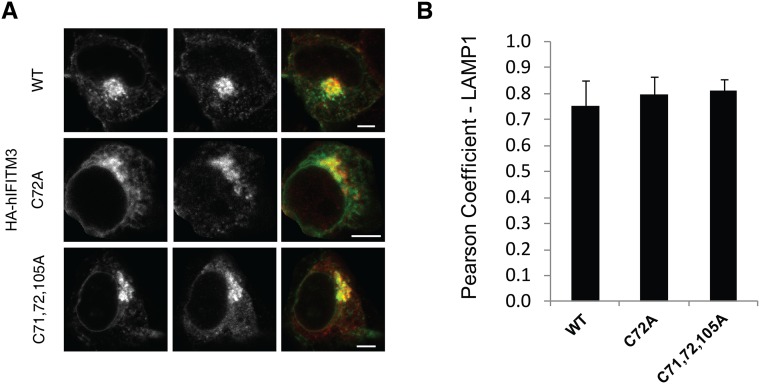
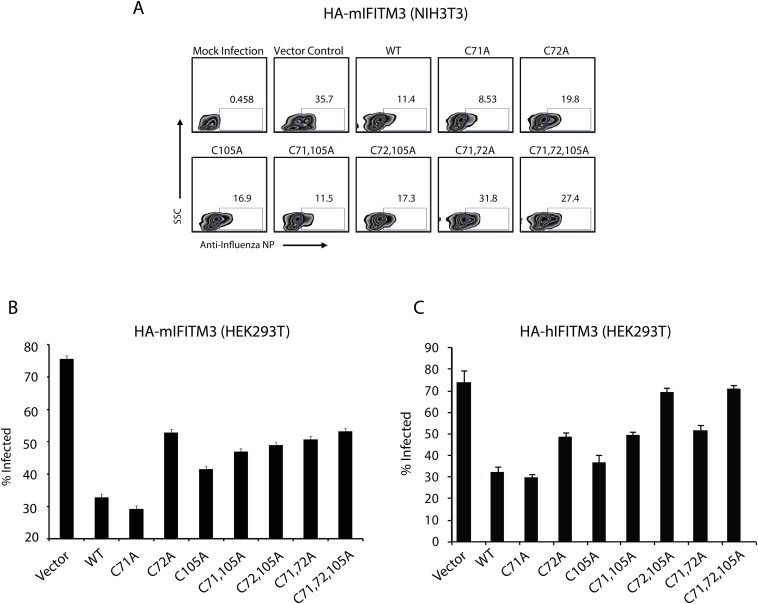
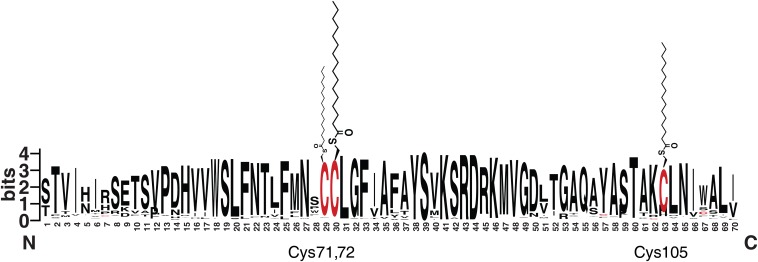
Fatty acylation of cysteine residues provides spatial and temporal control of protein function in cells and regulates important biological pathways in eukaryotes. Although recent methods have improved the detection and proteomic analysis of cysteine fatty (S-fatty) acylated proteins, understanding how specific sites and quantitative levels of this posttranslational modification modulate cellular pathways are still challenging. To analyze the endogenous levels of protein S-fatty acylation in cells, we developed a mass-tag labeling method based on hydroxylamine-sensitivity of thioesters and selective maleimide-modification of cysteines, termed acyl-PEG exchange (APE). We demonstrate that APE enables sensitive detection of protein S-acylation levels and is broadly applicable to different classes of S-palmitoylated membrane proteins. Using APE, we show that endogenous interferon-induced transmembrane protein 3 is S-fatty acylated on three cysteine residues and site-specific modification of highly conserved cysteines are crucial for the antiviral activity of this IFN-stimulated immune effector. APE therefore provides a general and sensitive method for analyzing the endogenous levels of protein S-fatty acylation and should facilitate quantitative studies of this regulated and dynamic lipid modification in biological systems.
Keywords: IFITM3; PEGylation; fatty-acylation; influenza virus; palmitoylation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Selective Enrichment and Direct Analysis of Protein S-Palmitoylation Sites.J Proteome Res. 2018 May 4;17(5):1907-1922. doi: 10.1021/acs.jproteome.8b00002. Epub 2018 Apr 6. J Proteome Res. 2018. PMID: 29575903 Free PMC article.
-
Mass-Tag Labeling Using Acyl-PEG Exchange for the Determination of Endogenous Protein S-Fatty Acylation.Curr Protoc Protein Sci. 2017 Aug 1;89:14.17.1-14.17.11. doi: 10.1002/cpps.36. Curr Protoc Protein Sci. 2017. PMID: 28762493 Free PMC article.
-
Chemical Proteomic Analysis of S-Fatty Acylated Proteins and Their Modification Sites.Methods Mol Biol. 2019;2009:45-57. doi: 10.1007/978-1-4939-9532-5_4. Methods Mol Biol. 2019. PMID: 31152394
-
Proteomic analysis of fatty-acylated proteins.Curr Opin Chem Biol. 2016 Feb;30:77-86. doi: 10.1016/j.cbpa.2015.11.008. Epub 2015 Dec 2. Curr Opin Chem Biol. 2016. PMID: 26656971 Free PMC article. Review.
-
The role of S-acylation in protein trafficking.Traffic. 2017 Nov;18(11):699-710. doi: 10.1111/tra.12510. Epub 2017 Sep 24. Traffic. 2017. PMID: 28837239 Review.
Cited by
-
Control of protein palmitoylation by regulating substrate recruitment to a zDHHC-protein acyltransferase.Commun Biol. 2020 Jul 31;3(1):411. doi: 10.1038/s42003-020-01145-3. Commun Biol. 2020. PMID: 32737405 Free PMC article.
-
Protein Lipidation Types: Current Strategies for Enrichment and Characterization.Int J Mol Sci. 2022 Feb 21;23(4):2365. doi: 10.3390/ijms23042365. Int J Mol Sci. 2022. PMID: 35216483 Free PMC article. Review.
-
S-Palmitoylation Sorts Membrane Cargo for Anterograde Transport in the Golgi.Dev Cell. 2018 Nov 19;47(4):479-493.e7. doi: 10.1016/j.devcel.2018.10.024. Dev Cell. 2018. PMID: 30458139 Free PMC article.
-
Covalent Chemical Tools for Profiling Post-Translational Modifications.Front Chem. 2022 Jul 4;10:868773. doi: 10.3389/fchem.2022.868773. eCollection 2022. Front Chem. 2022. PMID: 35860626 Free PMC article. Review.
-
Inhibition of protein N-myristoylation blocks Plasmodium falciparum intraerythrocytic development, egress and invasion.PLoS Biol. 2021 Oct 25;19(10):e3001408. doi: 10.1371/journal.pbio.3001408. eCollection 2021 Oct. PLoS Biol. 2021. PMID: 34695132 Free PMC article.
References
-
- Liang X, et al. Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J Biol Chem. 2001;276(33):30987–30994. - PubMed
-
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11(3):161–175. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
