

Heterotrimeric G-protein shuttling via Gip1 extends the dynamic range of eukaryotic chemotaxis
- PMID: 27044073
- PMCID: PMC4843477
- DOI: 10.1073/pnas.1516767113
Heterotrimeric G-protein shuttling via Gip1 extends the dynamic range of eukaryotic chemotaxis
Abstract
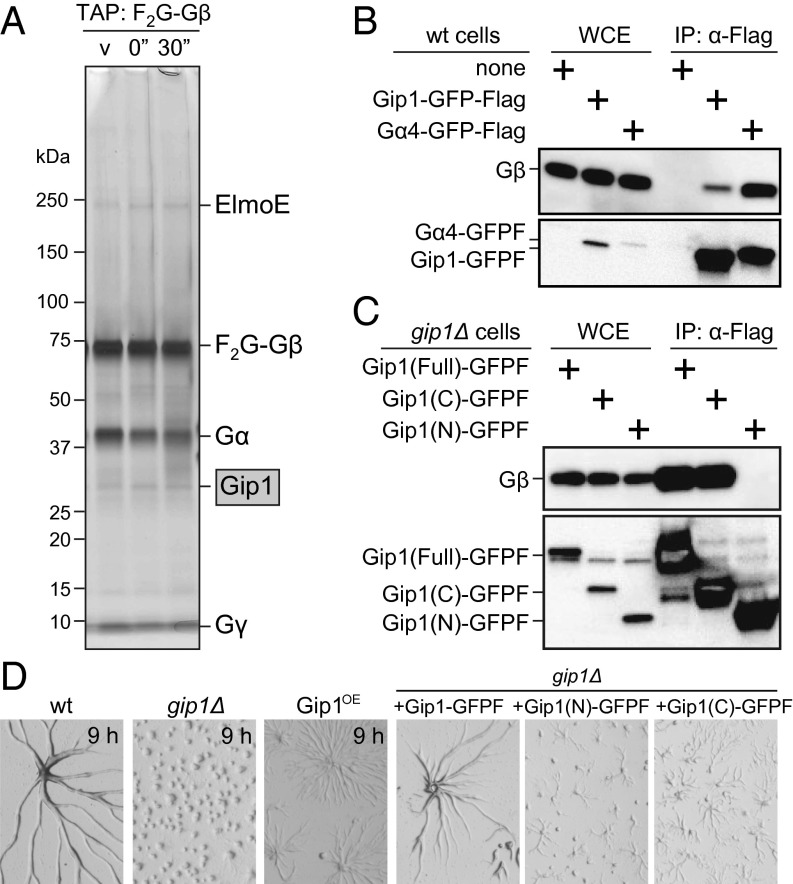
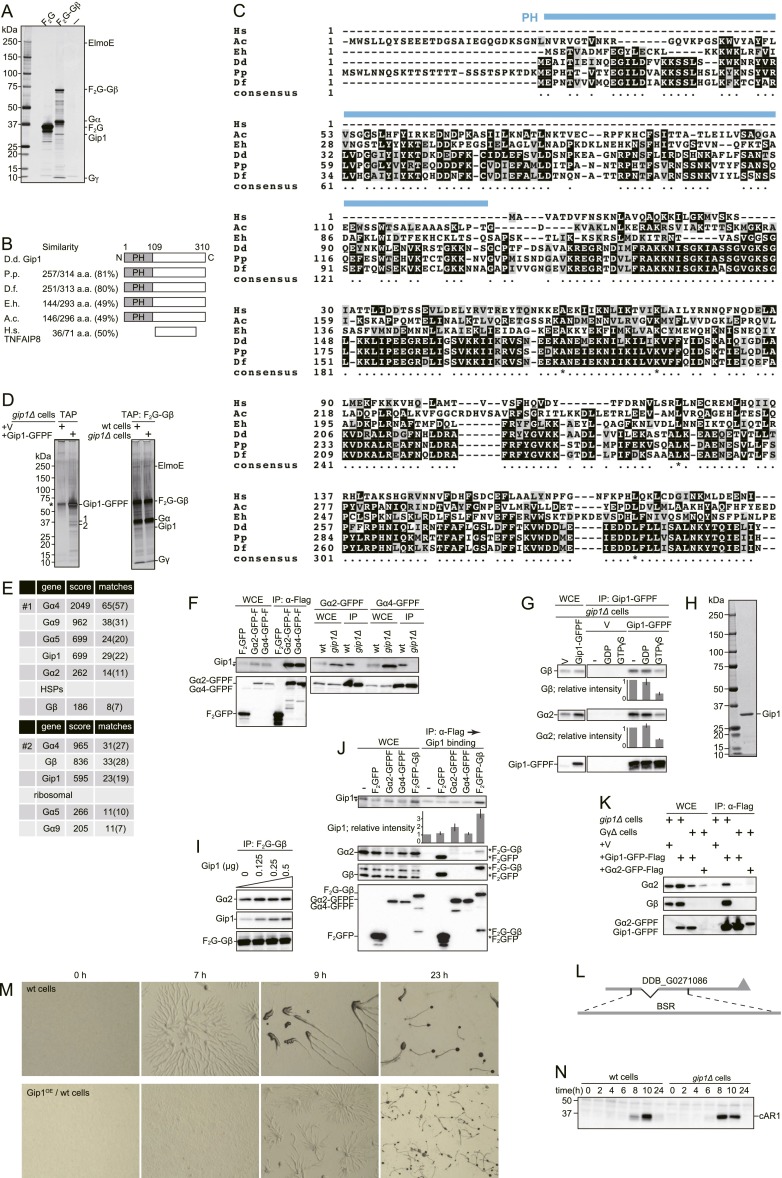
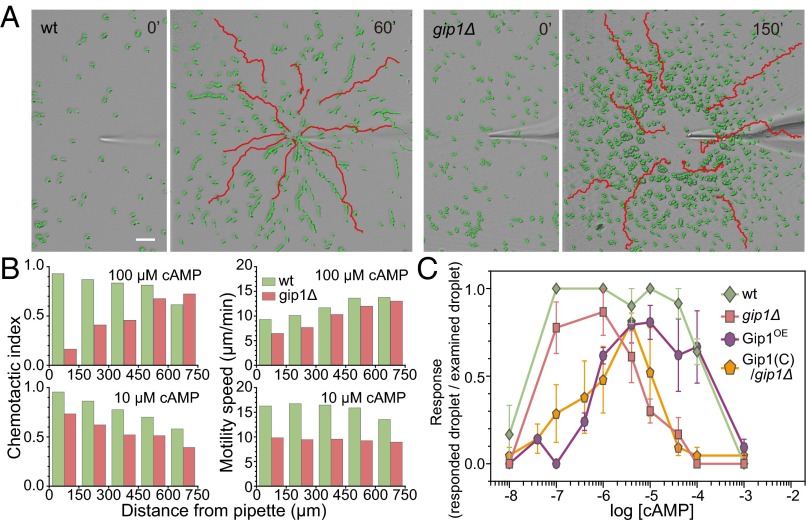
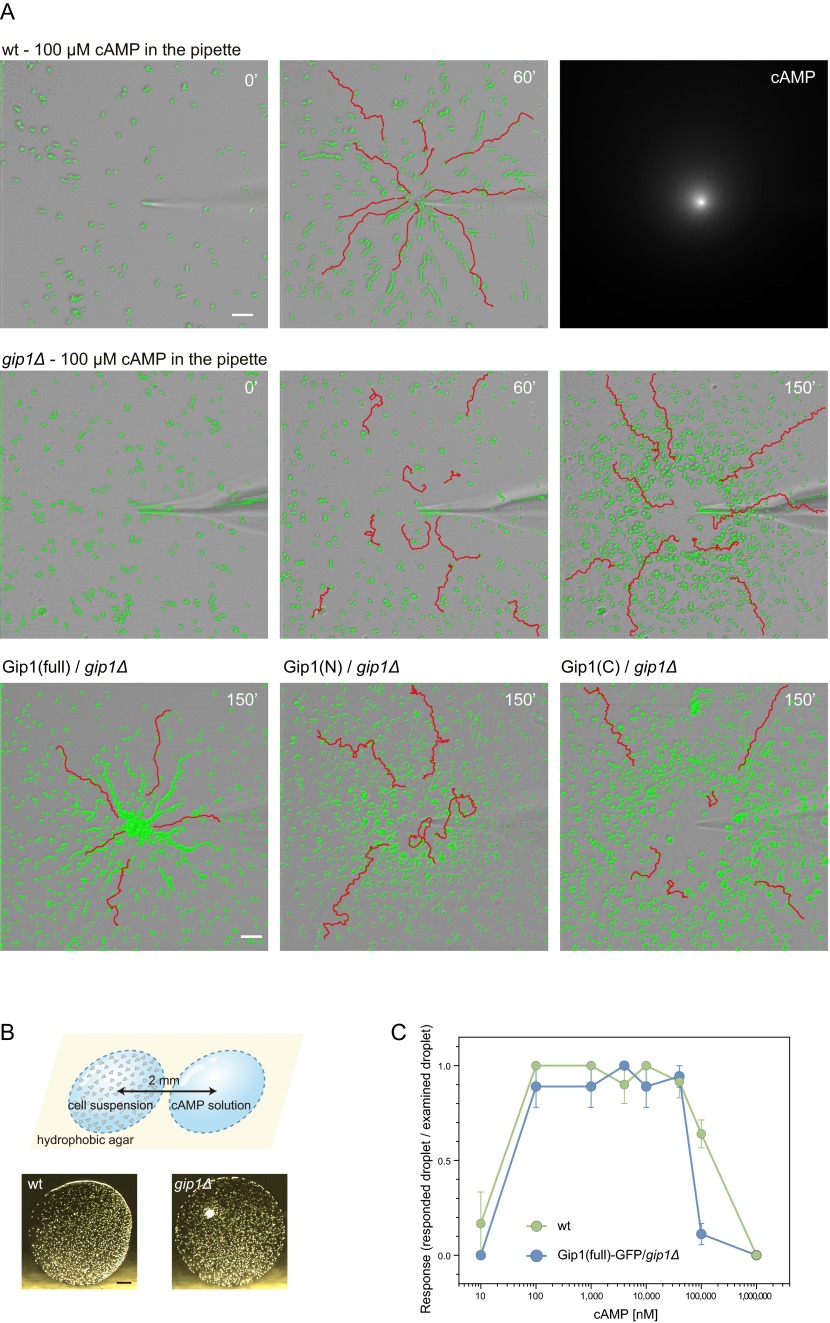
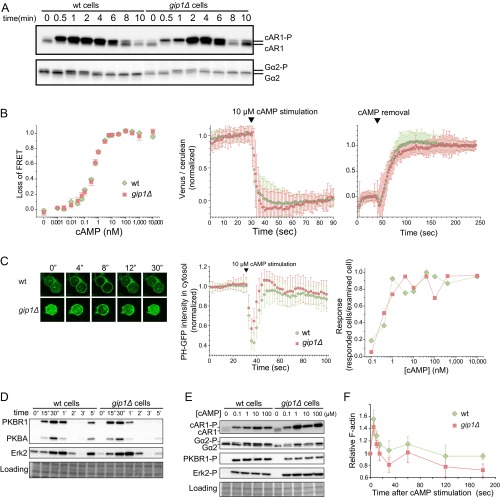
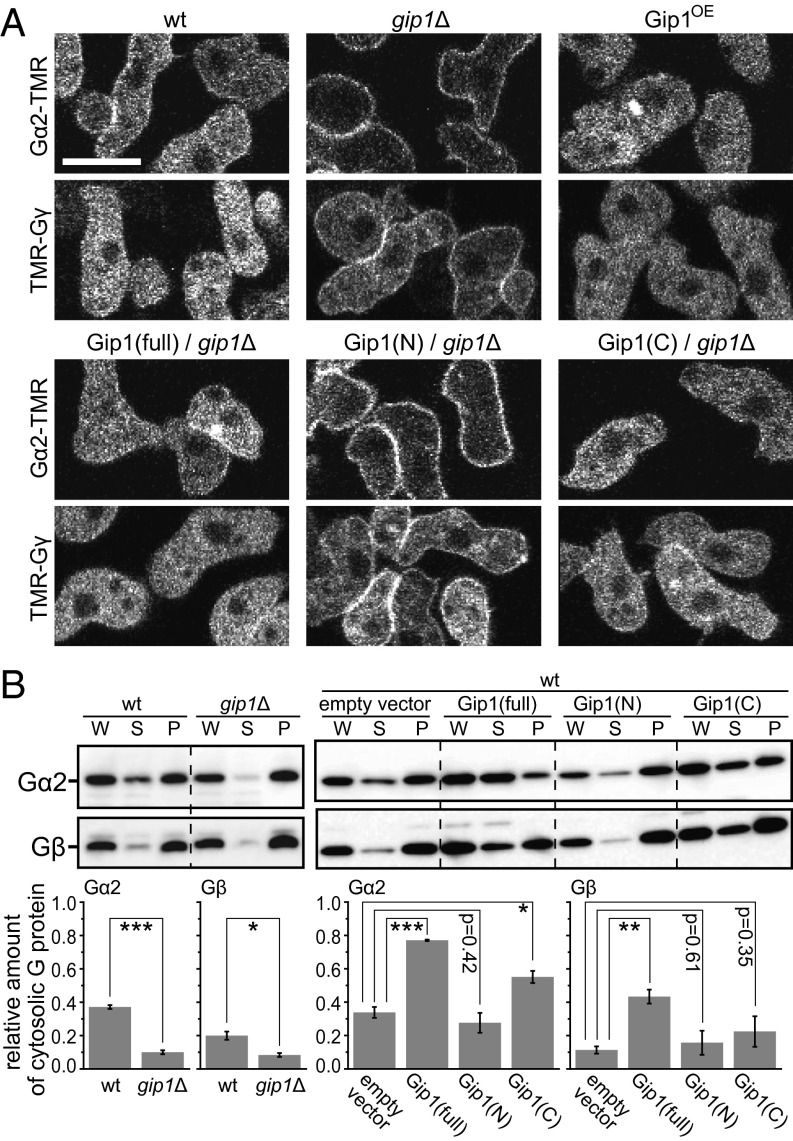
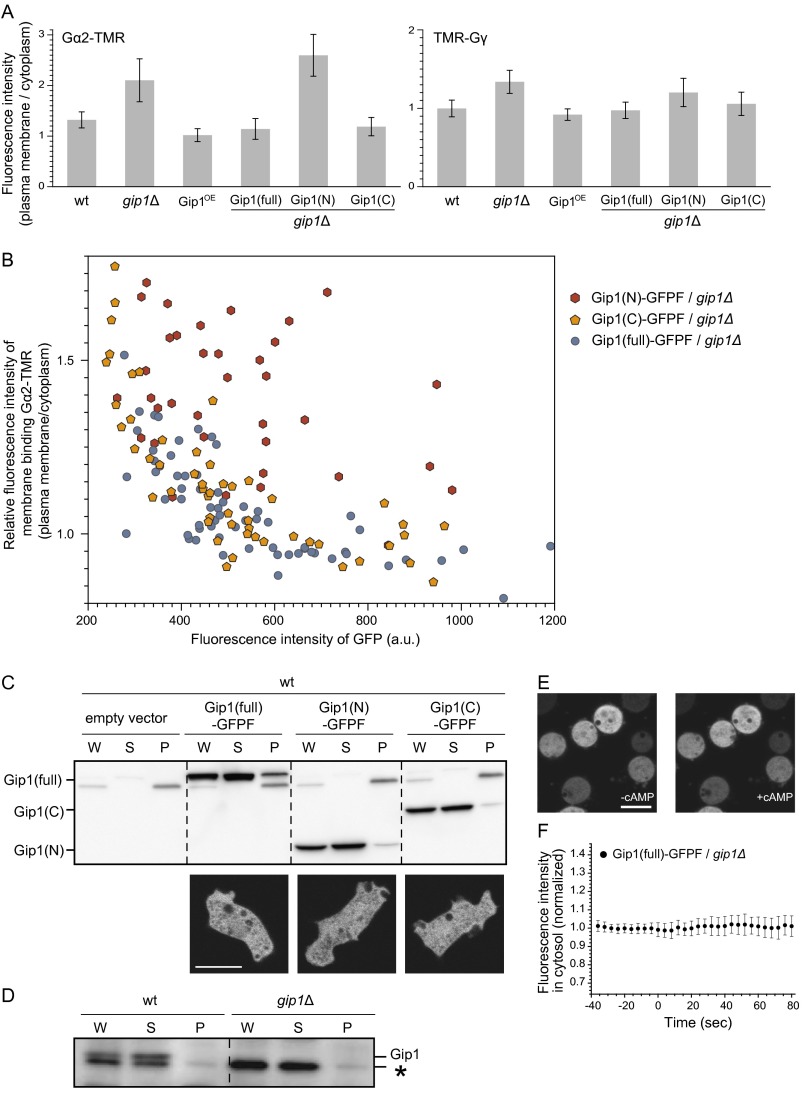
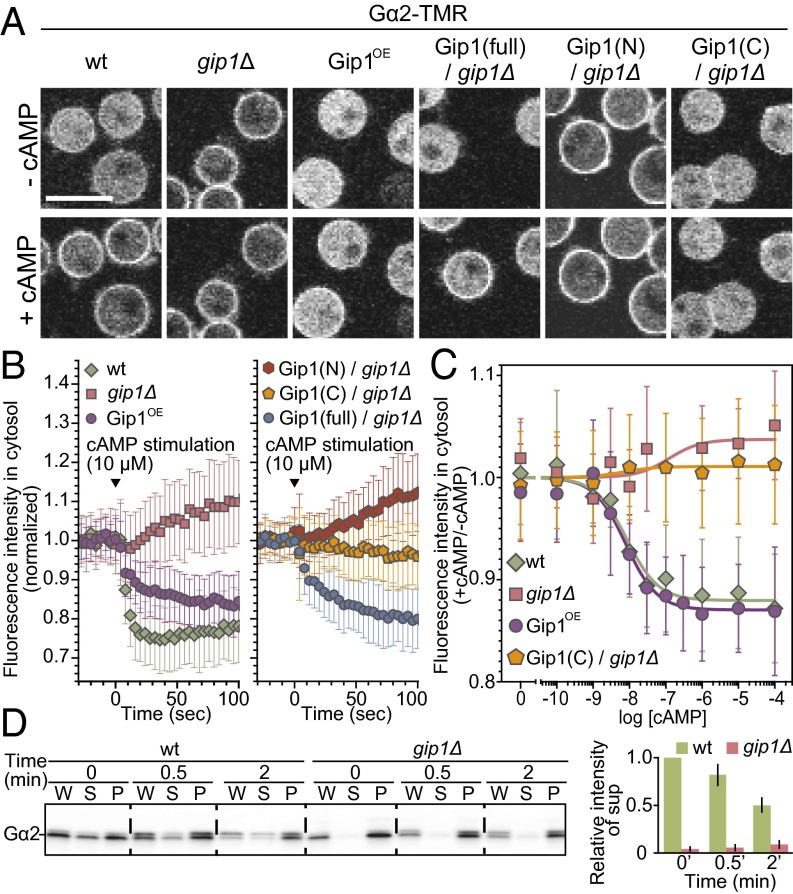
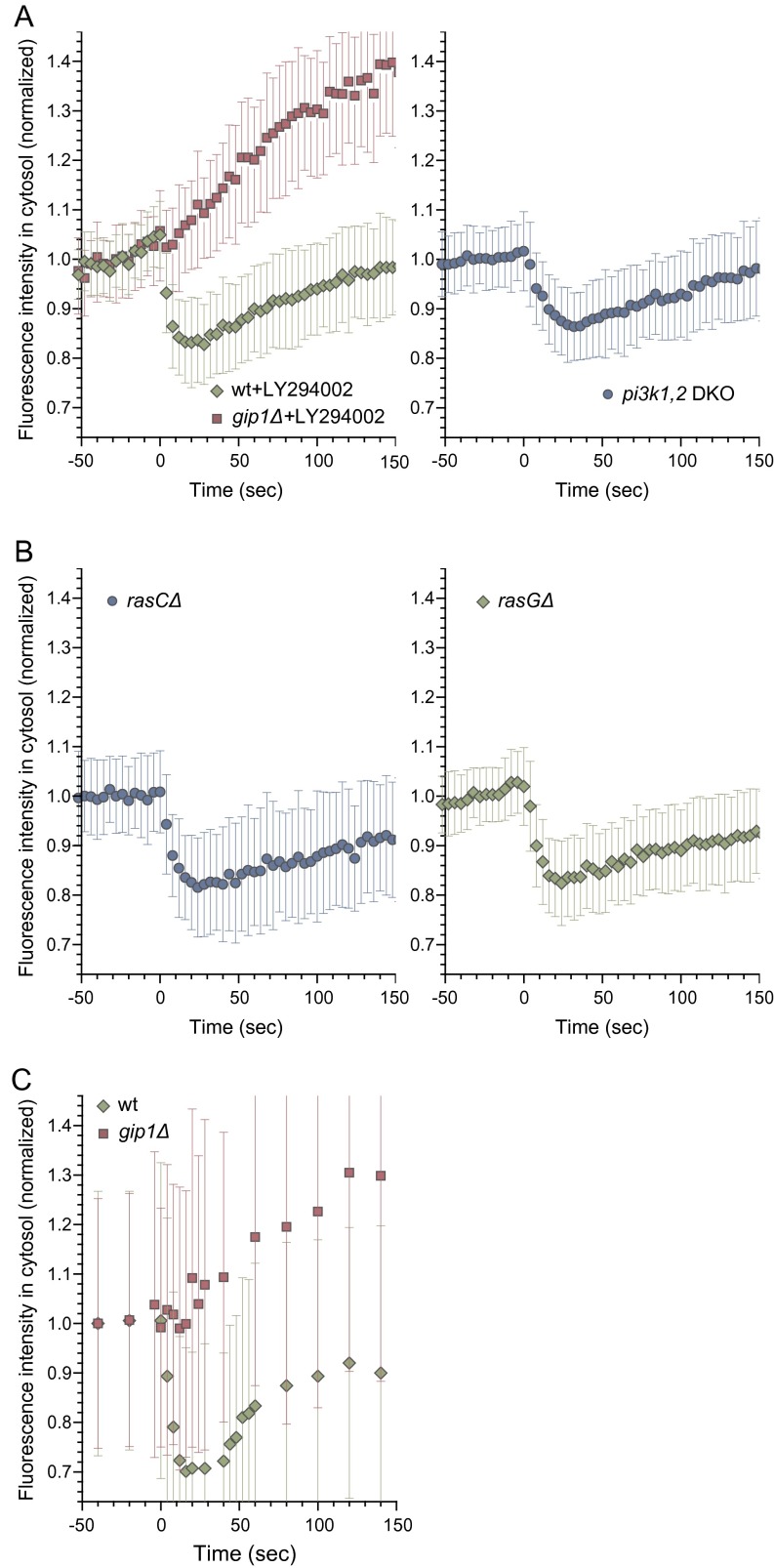
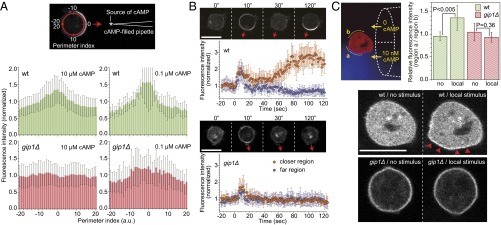
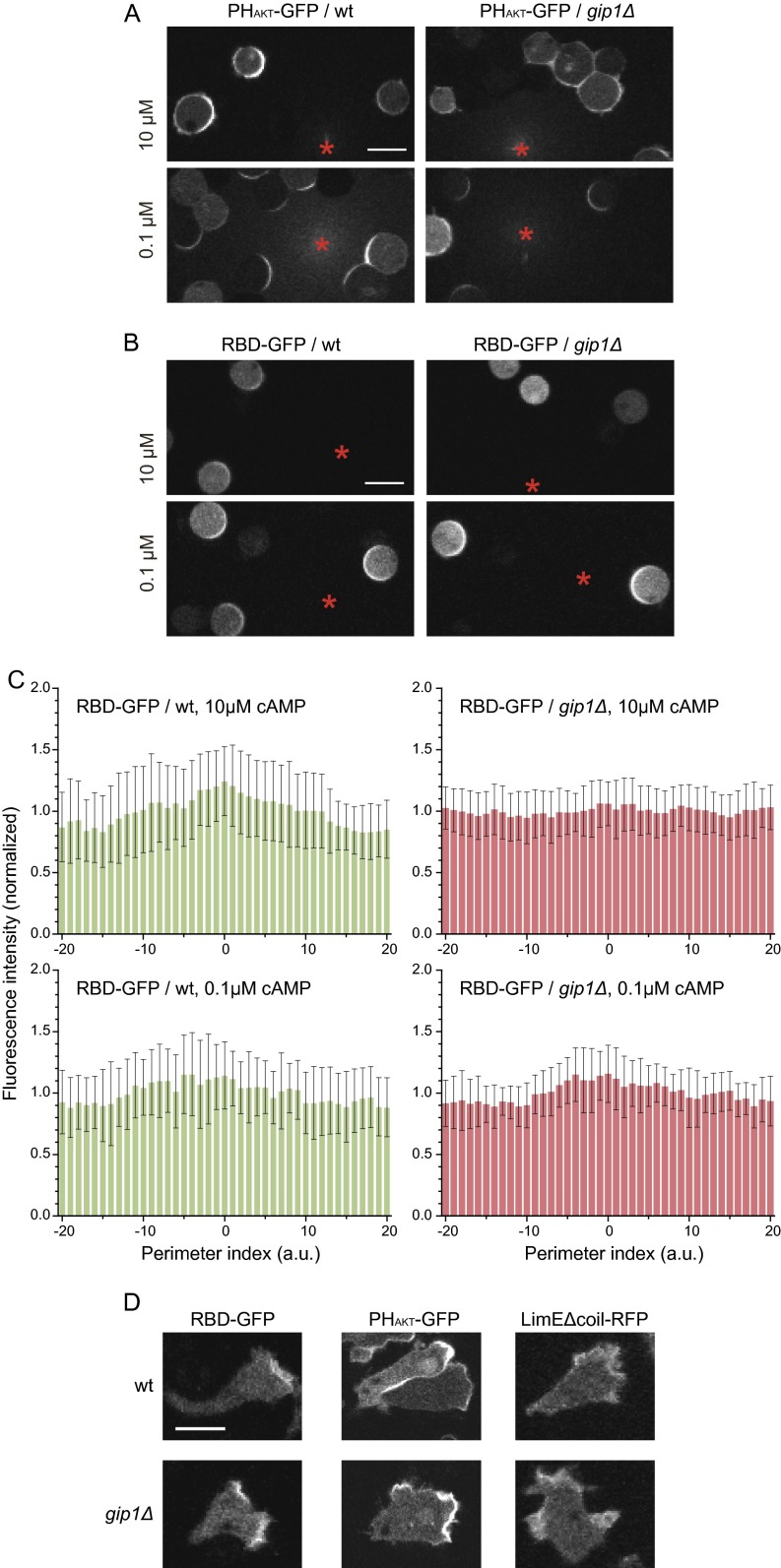
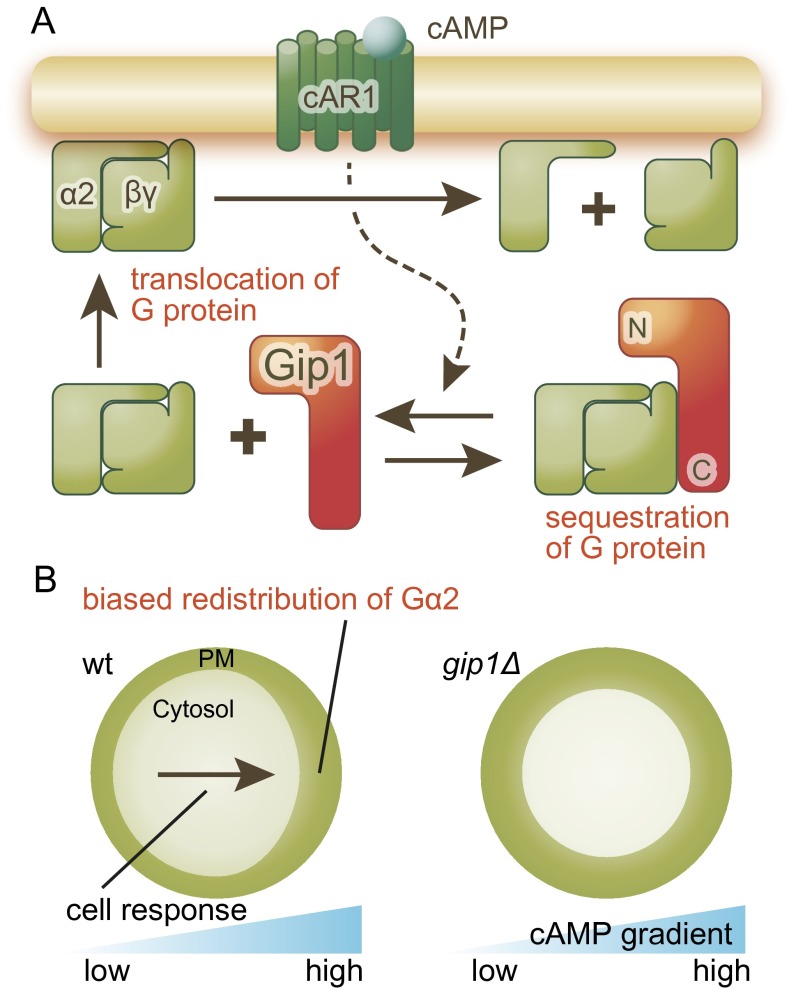
Chemotactic eukaryote cells can sense chemical gradients over a wide range of concentrations via heterotrimeric G-protein signaling; however, the underlying wide-range sensing mechanisms are only partially understood. Here we report that a novel regulator of G proteins, G protein-interacting protein 1 (Gip1), is essential for extending the chemotactic range ofDictyosteliumcells. Genetic disruption of Gip1 caused severe defects in gradient sensing and directed cell migration at high but not low concentrations of chemoattractant. Also, Gip1 was found to bind and sequester G proteins in cytosolic pools. Receptor activation induced G-protein translocation to the plasma membrane from the cytosol in a Gip1-dependent manner, causing a biased redistribution of G protein on the membrane along a chemoattractant gradient. These findings suggest that Gip1 regulates G-protein shuttling between the cytosol and the membrane to ensure the availability and biased redistribution of G protein on the membrane for receptor-mediated chemotactic signaling. This mechanism offers an explanation for the wide-range sensing seen in eukaryotic chemotaxis.
Keywords: dynamic range extension; eukaryotic chemotaxis; gradient sensing; heterotrimeric G protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Structural basis of Gip1 for cytosolic sequestration of G protein in wide-range chemotaxis.Nat Commun. 2018 Nov 6;9(1):4635. doi: 10.1038/s41467-018-07035-x. Nat Commun. 2018. PMID: 30401901 Free PMC article.
-
GPCR Signaling Regulation in Dictyostelium Chemotaxis.Methods Mol Biol. 2021;2274:317-336. doi: 10.1007/978-1-0716-1258-3_27. Methods Mol Biol. 2021. PMID: 34050483
-
Ggamma in dictyostelium: its role in localization of gbetagamma to the membrane is required for chemotaxis in shallow gradients.Mol Biol Cell. 2001 Oct;12(10):3204-13. doi: 10.1091/mbc.12.10.3204. Mol Biol Cell. 2001. PMID: 11598203 Free PMC article.
-
Function and Regulation of Heterotrimeric G Proteins during Chemotaxis.Int J Mol Sci. 2016 Jan 14;17(1):90. doi: 10.3390/ijms17010090. Int J Mol Sci. 2016. PMID: 26784171 Free PMC article. Review.
-
Gradient sensing during chemotaxis.Curr Opin Cell Biol. 2013 Oct;25(5):532-7. doi: 10.1016/j.ceb.2013.06.007. Epub 2013 Jul 20. Curr Opin Cell Biol. 2013. PMID: 23880435 Review.
Cited by
-
Spontaneous signal generation by an excitable system for cell migration.Front Cell Dev Biol. 2024 Feb 28;12:1373609. doi: 10.3389/fcell.2024.1373609. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 38481533 Free PMC article. Review.
-
GPCR-controlled membrane recruitment of negative regulator C2GAP1 locally inhibits Ras signaling for adaptation and long-range chemotaxis.Proc Natl Acad Sci U S A. 2017 Nov 21;114(47):E10092-E10101. doi: 10.1073/pnas.1703208114. Epub 2017 Nov 6. Proc Natl Acad Sci U S A. 2017. PMID: 29109256 Free PMC article.
-
The TIPE Molecular Pilot That Directs Lymphocyte Migration in Health and Inflammation.Sci Rep. 2020 Apr 20;10(1):6617. doi: 10.1038/s41598-020-63629-w. Sci Rep. 2020. PMID: 32313148 Free PMC article.
-
Field model for multistate lateral diffusion of various transmembrane proteins observed in living Dictyostelium cells.J Cell Sci. 2023 Feb 15;136(4):jcs260280. doi: 10.1242/jcs.260280. Epub 2023 Feb 20. J Cell Sci. 2023. PMID: 36655427 Free PMC article.
-
Proteomic and Transcriptomic Profiling Identifies Early Developmentally Regulated Proteins in Dictyostelium Discoideum.Cells. 2019 Oct 1;8(10):1187. doi: 10.3390/cells8101187. Cells. 2019. PMID: 31581556 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
