

An evolutionary roadmap to the microtubule-associated protein MAP Tau
- PMID: 27030133
- PMCID: PMC4815063
- DOI: 10.1186/s12864-016-2590-9
An evolutionary roadmap to the microtubule-associated protein MAP Tau
Abstract
Background: The microtubule associated protein Tau (MAPT) promotes assembly and interaction of microtubules with the cytoskeleton, impinging on axonal transport and synaptic plasticity. Its neuronal expression and intrinsic disorder implicate it in some 30 tauopathies such as Alzheimer's disease and frontotemporal dementia. These pathophysiological studies have yet to be complemented by computational analyses of its molecular evolution and structural models of all its functional domains to explain the molecular basis for its conservation profile, its site-specific interactions and the propensity to conformational disorder and aggregate formation.
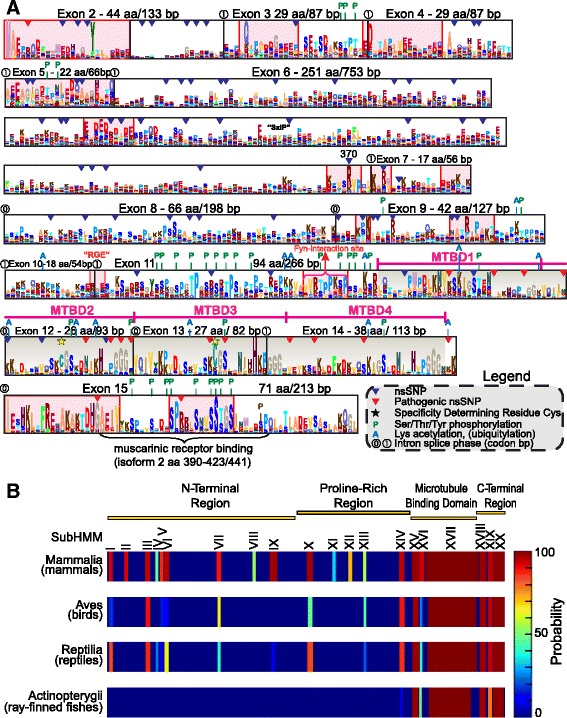
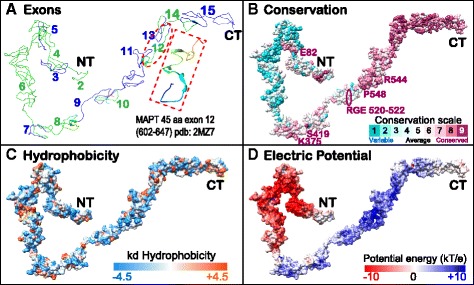
Results: We systematically annotated public sequence data to reconstruct unspliced MAPT, MAP2 and MAP4 transcripts spanning all represented genomes. Bayesian and maximum likelihood phylogenetic analyses, genetic linkage maps and domain architectures distinguished a nonvertebrate outgroup from the emergence of MAP4 and its subsequent ancestral duplication to MAP2 and MAPT. These events were coupled to other linked genes such as KANSL1L and KANSL and may thus be consequent to large-scale chromosomal duplications originating in the extant vertebrate genomes of hagfish and lamprey. Profile hidden Markov models (pHMMs), clustered subalignments and 3D structural predictions defined potential interaction motifs and specificity determining sites to reveal distinct signatures between the four homologous microtubule binding domains and independent divergence of the amino terminus.
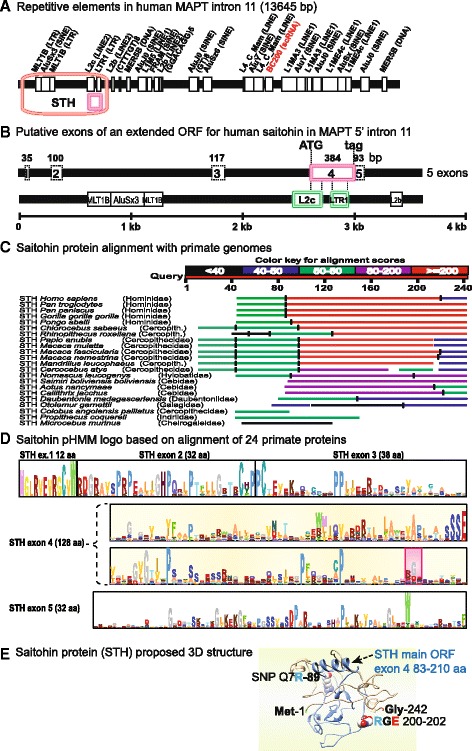
Conclusion: These analyses clarified ambiguities of MAPT nomenclature, defined the order, timing and pattern of its molecular evolution and identified key residues and motifs relevant to its protein interaction properties and pathogenic role. Additional unexpected findings included the expansion of cysteine-containing, microtubule binding domains of MAPT in cold adapted Antarctic icefish and the emergence of a novel multiexonic saitohin (STH) gene from repetitive elements in MAPT intron 11 of certain primate genomes.
Keywords: Domain architecture; Gene phylogeny; MAPT gene); Microtubule associated protein Tau (MAPT protein; Microtubule binding domain; Molecular evolution; Profile hidden Markov models; Saitohin (STH); Structure-function prediction.
Figures



Similar articles
-
Hypoxia alters expression of zebrafish microtubule-associated protein tau (mapta, maptb) gene transcripts.BMC Res Notes. 2014 Oct 31;7:767. doi: 10.1186/1756-0500-7-767. BMC Res Notes. 2014. PMID: 25359609 Free PMC article.
-
Much More Than a Cytoskeletal Protein: Physiological and Pathological Functions of the Non-microtubule Binding Region of Tau.Front Neurol. 2020 Oct 19;11:590059. doi: 10.3389/fneur.2020.590059. eCollection 2020. Front Neurol. 2020. PMID: 33193056 Free PMC article. Review.
-
Tau repeat regions contain conserved histidine residues that modulate microtubule-binding in response to changes in pH.J Biol Chem. 2019 May 31;294(22):8779-8790. doi: 10.1074/jbc.RA118.007004. Epub 2019 Apr 16. J Biol Chem. 2019. PMID: 30992364 Free PMC article.
-
[The genetics of dementias. Part 1: Molecular basis of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17)].Postepy Hig Med Dosw (Online). 2009 Jun 15;63:278-86. Postepy Hig Med Dosw (Online). 2009. PMID: 19535823 Review. Polish.
-
The MAP2/Tau family of microtubule-associated proteins.Genome Biol. 2005;6(1):204. doi: 10.1186/gb-2004-6-1-204. Epub 2004 Dec 23. Genome Biol. 2005. PMID: 15642108 Free PMC article. Review.
Cited by
-
Characterization of microtubule-associated protein tau isoforms and Alzheimer's disease-like pathology in normal sheep (Ovis aries): relevance to their potential as a model of Alzheimer's disease.Cell Mol Life Sci. 2022 Oct 21;79(11):560. doi: 10.1007/s00018-022-04572-z. Cell Mol Life Sci. 2022. PMID: 36269420 Free PMC article.
-
Microtubule-modulating Agents in the Fight Against Neurodegeneration: Will it ever Work?Curr Neuropharmacol. 2022;20(4):782-798. doi: 10.2174/1570159X19666211201101020. Curr Neuropharmacol. 2022. PMID: 34852744 Free PMC article. Review.
-
Propagation of Tau aggregates.Mol Brain. 2017 May 30;10(1):18. doi: 10.1186/s13041-017-0298-7. Mol Brain. 2017. PMID: 28558799 Free PMC article. Review.
-
Characterization of Intrinsically Disordered Proteins in Healthy and Diseased States by Nuclear Magnetic Resonance.Rev Recent Clin Trials. 2024;19(3):176-188. doi: 10.2174/0115748871271420240213064251. Rev Recent Clin Trials. 2024. PMID: 38409704 Review.
-
The Evolution of Tau Phosphorylation and Interactions.Front Aging Neurosci. 2019 Sep 18;11:256. doi: 10.3389/fnagi.2019.00256. eCollection 2019. Front Aging Neurosci. 2019. PMID: 31619983 Free PMC article.
References
-
- Derisbourg M, Leghay C, Chiapetta G, Fernandez-Gomez FJ, Laurent C, Demeyer D, Carrier S, Buée-Scherrer V, Blum D, Vinh J, Sergeant N, Verdier Y, Buée L, Hamdane M. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep. 2015;5:9659. doi: 10.1038/srep09659. - DOI - PMC - PubMed
-
- Fichou Y, Heyden M, Zaccai G, Weik M, Tobias DJ. Molecular dynamics simulations of a powder model of the intrinsically disordered protein Tau. J Phys Chem B. 2015; doi:10.1021/acs.jpcb.5b05849 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
