

Targeting metastasis
- PMID: 27009393
- PMCID: PMC7055530
- DOI: 10.1038/nrc.2016.25
Targeting metastasis
Abstract
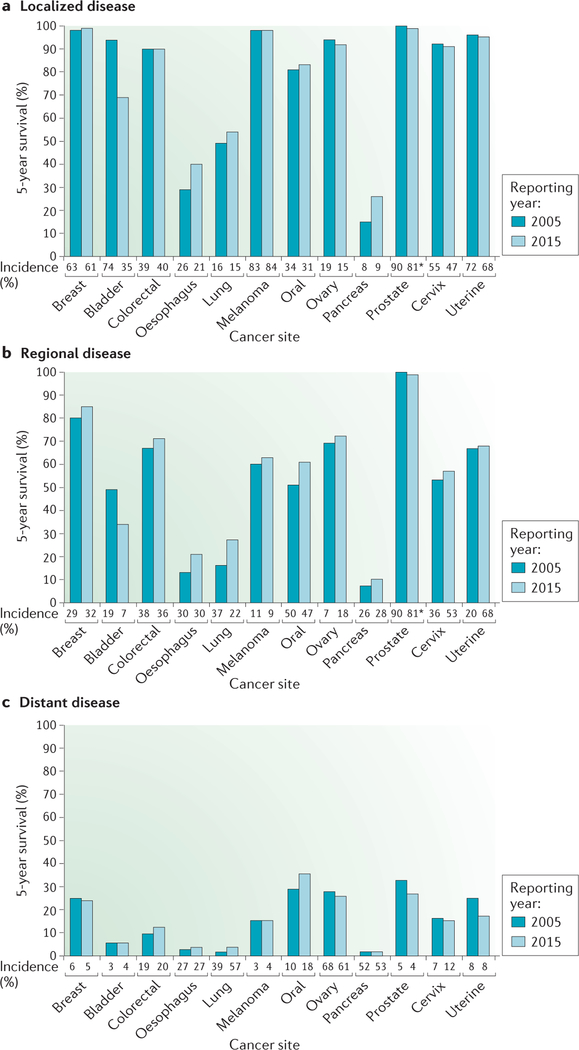
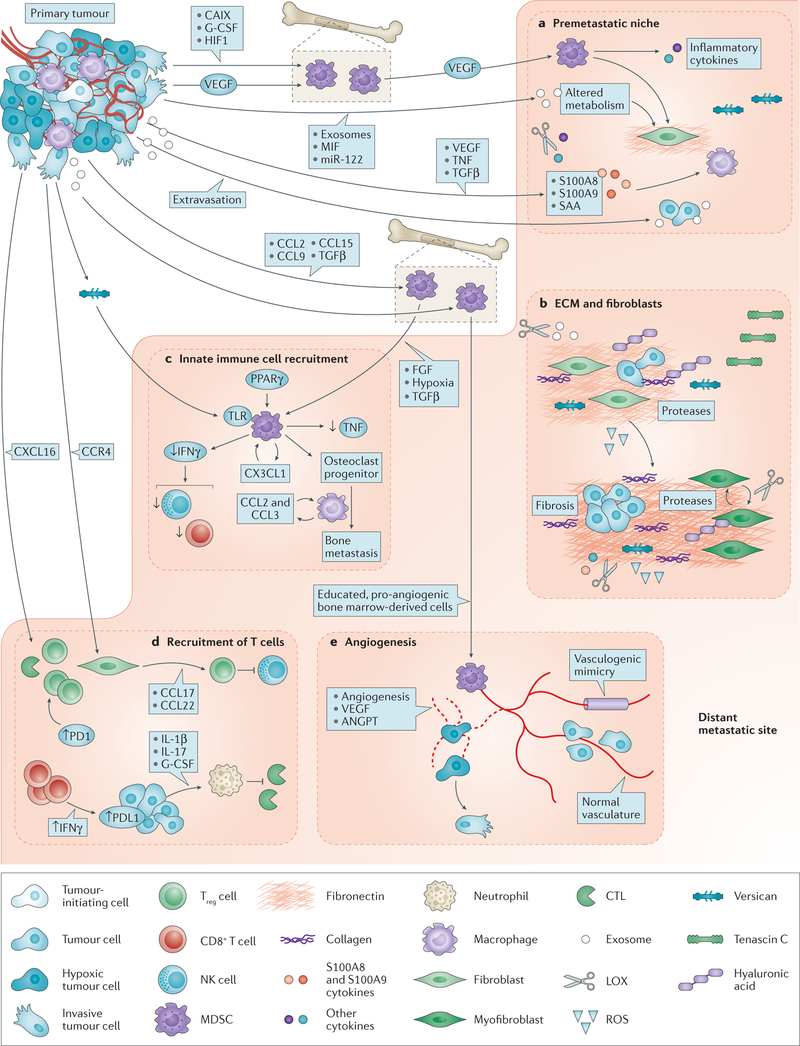
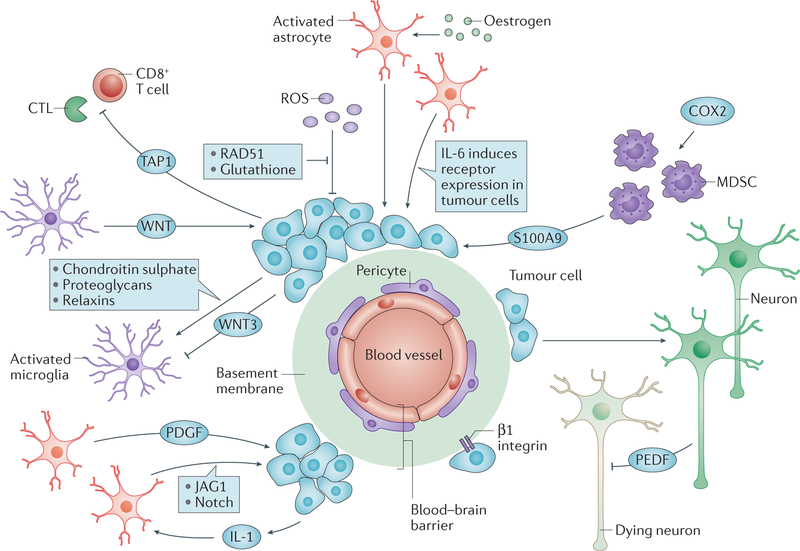
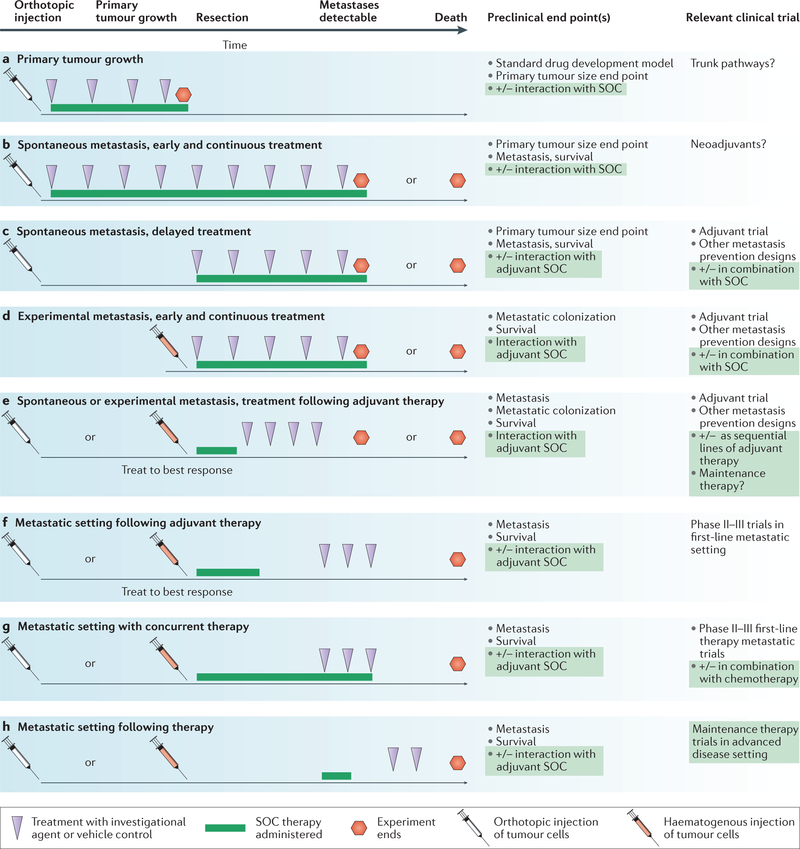
Tumour metastasis, the movement of tumour cells from a primary site to progressively colonize distant organs, is a major contributor to the deaths of cancer patients. Therapeutic goals are the prevention of an initial metastasis in high-risk patients, shrinkage of established lesions and prevention of additional metastases in patients with limited disease. Instead of being autonomous, tumour cells engage in bidirectional interactions with metastatic microenvironments to alter antitumour immunity, the extracellular milieu, genomic stability, survival signalling, chemotherapeutic resistance and proliferative cycles. Can targeting of these interactions significantly improve patient outcomes? In this Review preclinical research, combination therapies and clinical trial designs are re-examined.
Conflict of interest statement
Competing interests statement
The authors declare
Figures
Similar articles
-
Current and emerging concepts in tumour metastasis.J Pathol. 2010 Sep;222(1):1-15. doi: 10.1002/path.2727. J Pathol. 2010. PMID: 20681009 Review.
-
Metastatic pathways and time courses in the orderly progression of cutaneous melanoma.Br J Dermatol. 2002 Jul;147(1):62-70. doi: 10.1046/j.1365-2133.2002.04867.x. Br J Dermatol. 2002. PMID: 12100186
-
Metastasis: the deadly part of cancer.Br J Nurs. 1996 May 9-22;5(9):535-8. doi: 10.12968/bjon.1996.5.9.535. Br J Nurs. 1996. PMID: 8716476 Review.
-
Inhibitors for metastasis development.Recent Pat Anticancer Drug Discov. 2006 Jan;1(1):69-80. doi: 10.2174/157489206775246511. Recent Pat Anticancer Drug Discov. 2006. PMID: 18221027 Review.
-
Clinical aspects of metastases.Ciba Found Symp. 1988;141:223-43. doi: 10.1002/9780470513736.ch13. Ciba Found Symp. 1988. PMID: 3075936 Review.
Cited by
-
Unveiling the therapeutic potential: KBU2046 halts triple-negative breast cancer cell migration by constricting TGF-β1 activation in vitro.Oncol Res. 2024 Oct 16;32(11):1791-1802. doi: 10.32604/or.2024.049348. eCollection 2024. Oncol Res. 2024. PMID: 39449805 Free PMC article.
-
In vivo library screening identifies the metabolic enzyme aldolase A as a promoter of metastatic lung colonization.iScience. 2021 Apr 20;24(5):102425. doi: 10.1016/j.isci.2021.102425. eCollection 2021 May 21. iScience. 2021. PMID: 34036247 Free PMC article.
-
Mechanoresponsive metabolism in cancer cell migration and metastasis.Cell Metab. 2021 Jul 6;33(7):1307-1321. doi: 10.1016/j.cmet.2021.04.002. Epub 2021 Apr 28. Cell Metab. 2021. PMID: 33915111 Free PMC article. Review.
-
Computer-Aided Intra-Operatory Positioning of an MRgHIFU Applicator Dedicated to Abdominal Thermal Therapy Using Particle Swarm Optimization.IEEE Open J Eng Med Biol. 2024 Jun 5;5:524-533. doi: 10.1109/OJEMB.2024.3410118. eCollection 2024. IEEE Open J Eng Med Biol. 2024. PMID: 39050977 Free PMC article.
-
Pan-cancer analysis reveals TAp63-regulated oncogenic lncRNAs that promote cancer progression through AKT activation.Nat Commun. 2020 Oct 14;11(1):5156. doi: 10.1038/s41467-020-18973-w. Nat Commun. 2020. PMID: 33056990 Free PMC article.
References
-
- Paget S The distribution of secondary growths in cancer of the breast. Lancet 1, 99–101 (1889). - PubMed
-
The origin of the seed and soil hypothesis of metastasis.
-
- Hensel JA, Flaig TW & Theodorescu D Clinical opportunities and challenges in targeting tumour dormancy. Nat. Rev. Clin. Oncol 10, 41–51 (2013). - PubMed
-
- Husemann Y et al. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68 (2008). - PubMed
-
- Bojovic B & Crowe DL Dysfunctional telomeres promote genomic instability and metastasis in the absence of telomerase activity in oncogene induced mammary cancer. Mol. Carcinogen 52, 103–117 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
