

Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1)
- PMID: 27002158
- PMCID: PMC4865928
- DOI: 10.1074/jbc.M115.698779
Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1)
Abstract
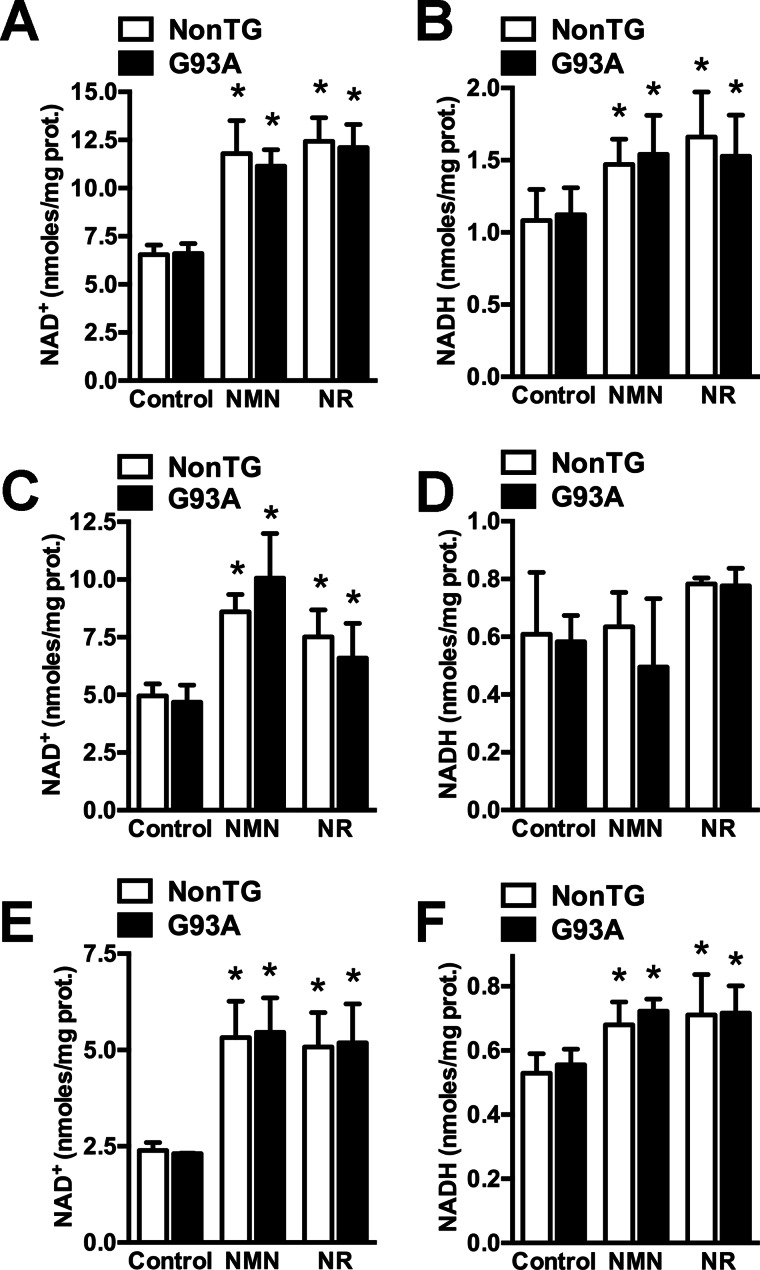
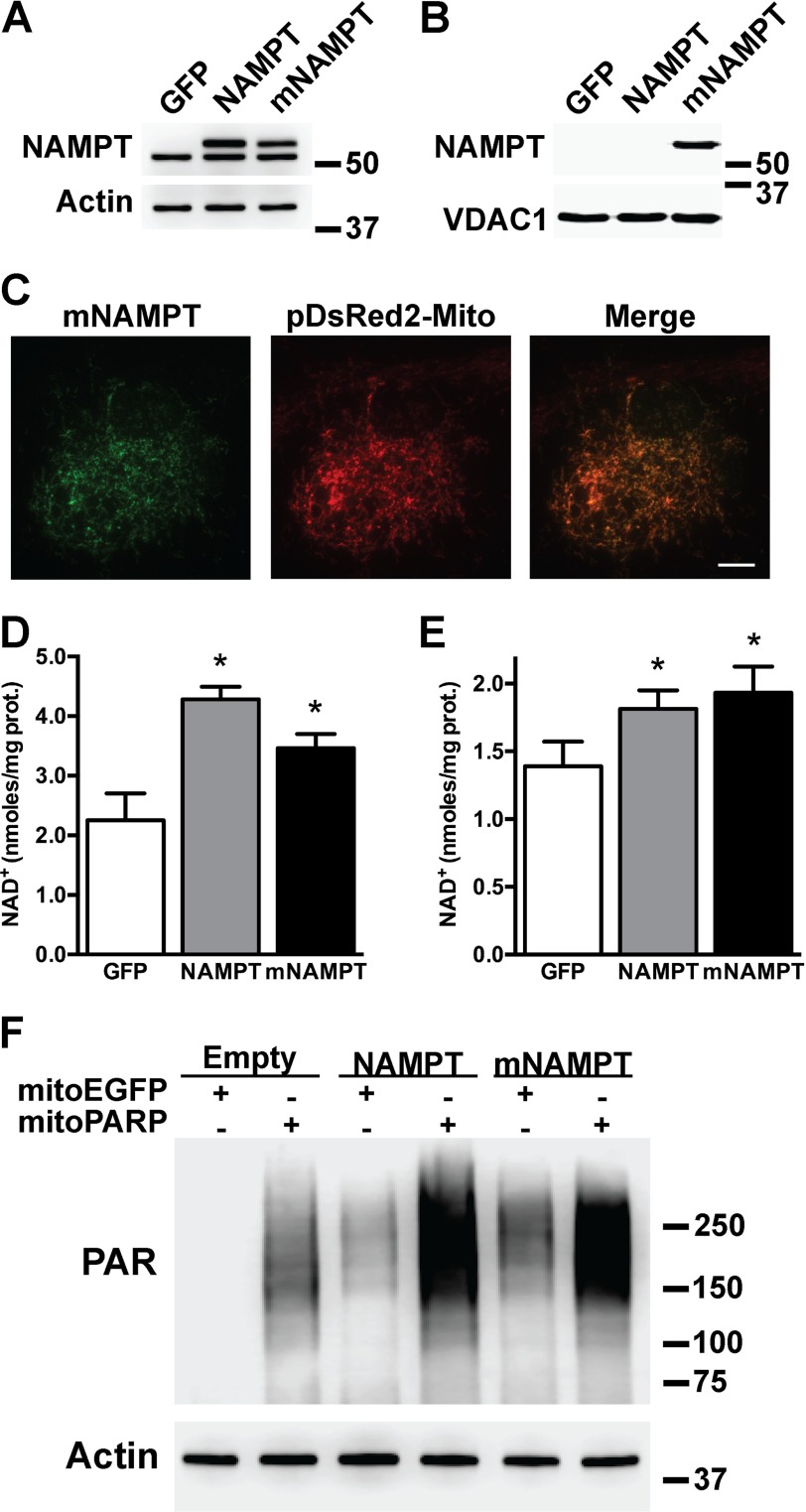
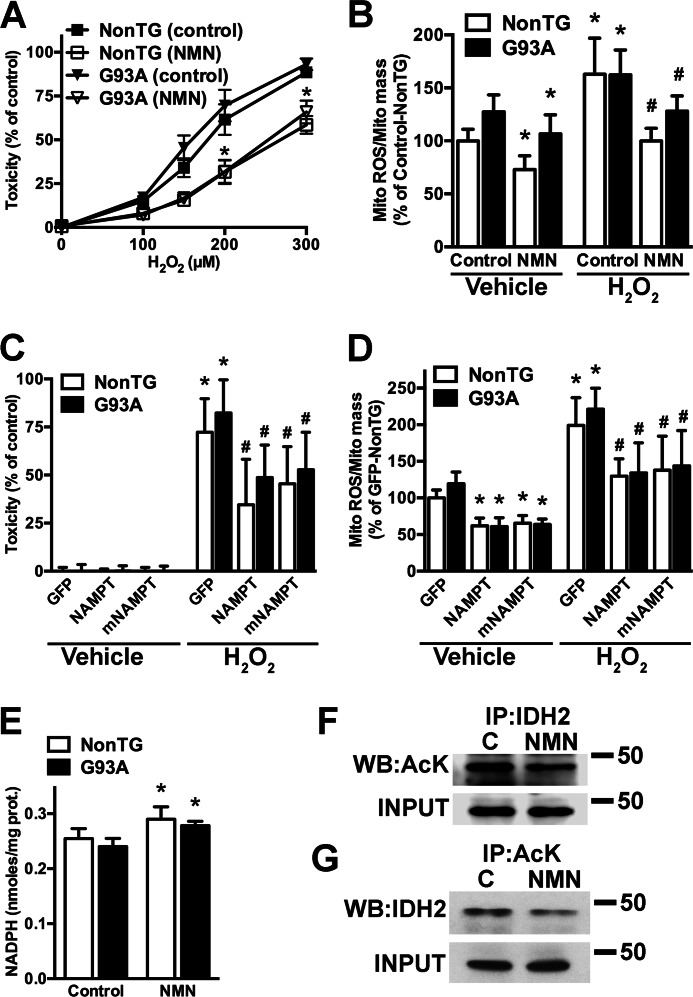
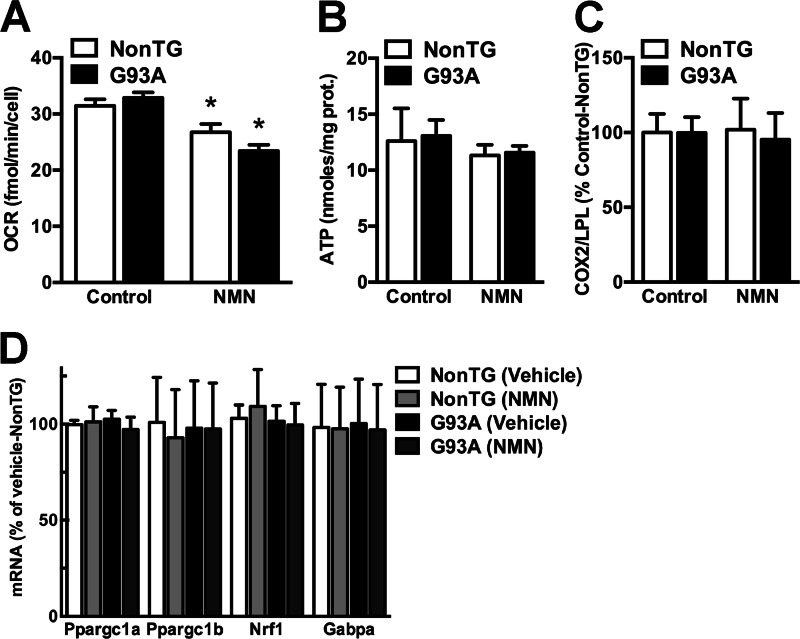
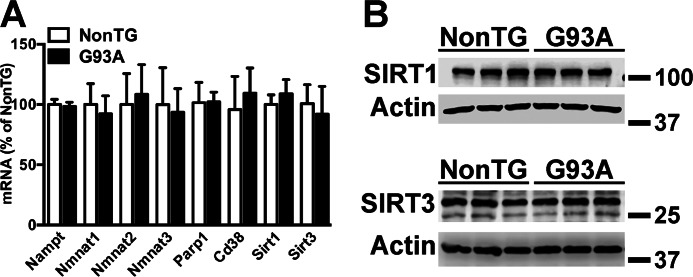
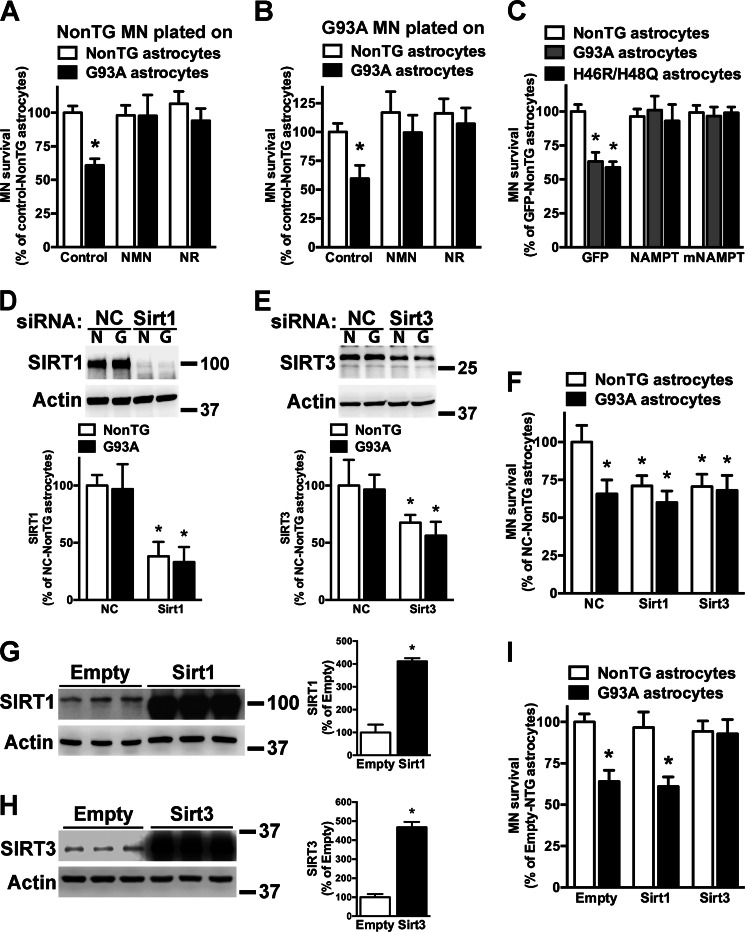
Nicotinamide adenine dinucleotide (NAD(+)) participates in redox reactions and NAD(+)-dependent signaling pathways. Although the redox reactions are critical for efficient mitochondrial metabolism, they are not accompanied by any net consumption of the nucleotide. On the contrary, NAD(+)-dependent signaling processes lead to its degradation. Three distinct families of enzymes consume NAD(+) as substrate: poly(ADP-ribose) polymerases, ADP-ribosyl cyclases (CD38 and CD157), and sirtuins (SIRT1-7). Because all of the above enzymes generate nicotinamide as a byproduct, mammalian cells have evolved an NAD(+) salvage pathway capable of resynthesizing NAD(+) from nicotinamide. Overexpression of the rate-limiting enzyme in this pathway, nicotinamide phosphoribosyltransferase, increases total and mitochondrial NAD(+) levels in astrocytes. Moreover, targeting nicotinamide phosphoribosyltransferase to the mitochondria also enhances NAD(+) salvage pathway in astrocytes. Supplementation with the NAD(+) precursors nicotinamide mononucleotide and nicotinamide riboside also increases NAD(+) levels in astrocytes. Amyotrophic lateral sclerosis (ALS) is caused by the progressive degeneration of motor neurons in the spinal cord, brain stem, and motor cortex. Superoxide dismutase 1 (SOD1) mutations account for up to 20% of familial ALS and 1-2% of apparently sporadic ALS cases. Primary astrocytes isolated from mutant human superoxide dismutase 1-overexpressing mice as well as human post-mortem ALS spinal cord-derived astrocytes induce motor neuron death in co-culture. Increasing total and mitochondrial NAD(+) content in ALS astrocytes increases oxidative stress resistance and reverts their toxicity toward co-cultured motor neurons. Taken together, our results suggest that enhancing the NAD(+) salvage pathway in astrocytes could be a potential therapeutic target to prevent astrocyte-mediated motor neuron death in ALS.
Keywords: NAD biosynthesis; NAMPT; amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease); astrocyte; mitochondria; oxidative stress.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Nicotinamide Adenine Dinucleotide Precursor Supplementation Modulates Neurite Complexity and Survival in Motor Neurons from Amyotrophic Lateral Sclerosis Models.Antioxid Redox Signal. 2024 Sep;41(7-9):573-589. doi: 10.1089/ars.2023.0360. Epub 2024 Jul 8. Antioxid Redox Signal. 2024. PMID: 38504592
-
Evaluation of the NAD+ biosynthetic pathway in ALS patients and effect of modulating NAD+ levels in hSOD1-linked ALS mouse models.Exp Neurol. 2020 May;327:113219. doi: 10.1016/j.expneurol.2020.113219. Epub 2020 Jan 31. Exp Neurol. 2020. PMID: 32014438 Free PMC article.
-
Mitochondria-targeted catalase reverts the neurotoxicity of hSOD1G⁹³A astrocytes without extending the survival of ALS-linked mutant hSOD1 mice.PLoS One. 2014 Jul 23;9(7):e103438. doi: 10.1371/journal.pone.0103438. eCollection 2014. PLoS One. 2014. PMID: 25054289 Free PMC article.
-
Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis.Neurodegener Dis. 2005;2(3-4):139-46. doi: 10.1159/000089619. Neurodegener Dis. 2005. PMID: 16909019 Review.
-
Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation.Neuropathology. 2001 Mar;21(1):82-92. doi: 10.1046/j.1440-1789.2001.00361.x. Neuropathology. 2001. PMID: 11304046 Review.
Cited by
-
Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis.Hum Mol Genet. 2018 Nov 1;27(21):3761-3771. doi: 10.1093/hmg/ddy260. Hum Mol Genet. 2018. PMID: 30010873 Free PMC article.
-
Mitochondrial dysregulation occurs early in ALS motor cortex with TDP-43 pathology and suggests maintaining NAD+ balance as a therapeutic strategy.Sci Rep. 2022 Mar 11;12(1):4287. doi: 10.1038/s41598-022-08068-5. Sci Rep. 2022. PMID: 35277554 Free PMC article.
-
Nicotinamide Adenine Dinucleotide Precursor Supplementation Modulates Neurite Complexity and Survival in Motor Neurons from Amyotrophic Lateral Sclerosis Models.Antioxid Redox Signal. 2024 Sep;41(7-9):573-589. doi: 10.1089/ars.2023.0360. Epub 2024 Jul 8. Antioxid Redox Signal. 2024. PMID: 38504592
-
Novel multifunctional iron chelators of the aroyl nicotinoyl hydrazone class that markedly enhance cellular NAD+ /NADH ratios.Br J Pharmacol. 2020 May;177(9):1967-1987. doi: 10.1111/bph.14963. Epub 2020 Feb 12. Br J Pharmacol. 2020. PMID: 31895471 Free PMC article.
-
Increased ROS Level in Spinal Cord of Wobbler Mice due to Nmnat2 Downregulation.Mol Neurobiol. 2018 Nov;55(11):8414-8424. doi: 10.1007/s12035-018-0999-7. Epub 2018 Mar 16. Mol Neurobiol. 2018. PMID: 29549647
References
-
- Berger F., Ramírez-Hernández M. H., and Ziegler M. (2004) The new life of a centenarian: signalling functions of NAD(P). Trends Biochem. Sci. 29, 111–118 - PubMed
-
- Bai P. (2015) Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol. Cell 58, 947–958 - PubMed
-
- Aksoy P., White T. A., Thompson M., and Chini E. N. (2006) Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 345, 1386–1392 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
