

Cross Talk between Nucleotide Synthesis Pathways with Cellular Immunity in Constraining Hepatitis E Virus Replication
- PMID: 26926637
- PMCID: PMC4862450
- DOI: 10.1128/AAC.02700-15
Cross Talk between Nucleotide Synthesis Pathways with Cellular Immunity in Constraining Hepatitis E Virus Replication
Abstract
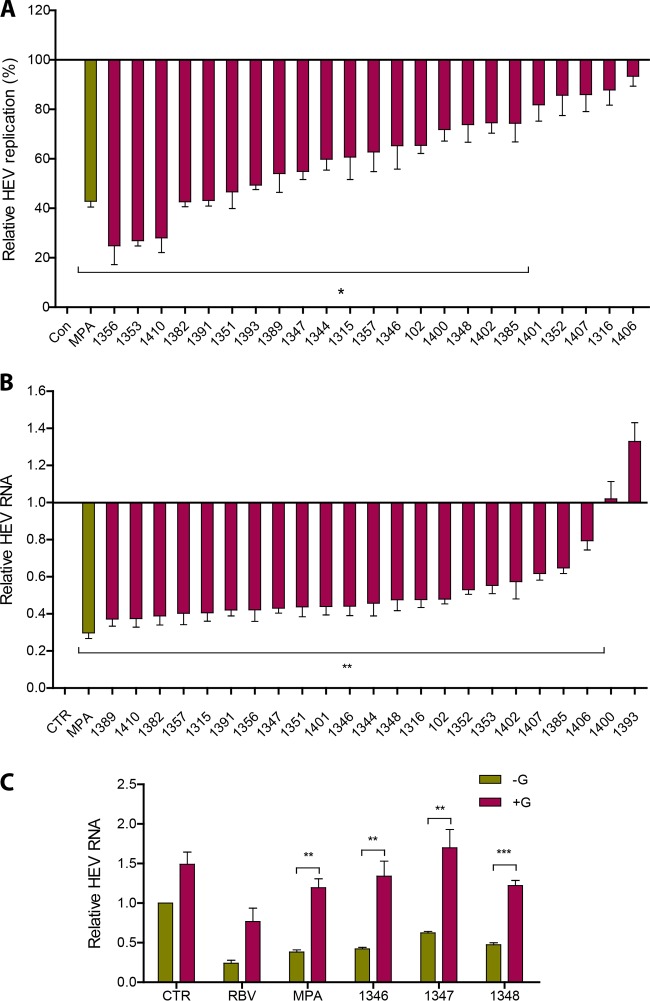
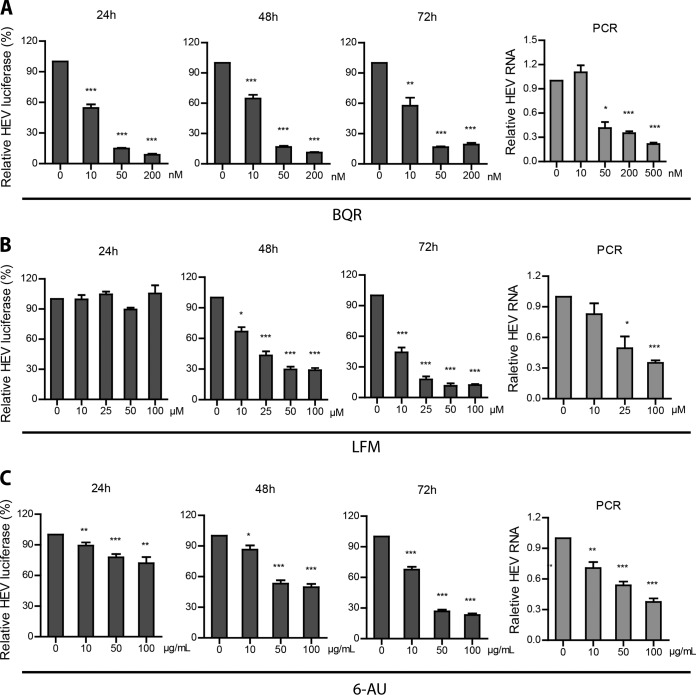
Viruses are solely dependent on host cells to propagate; therefore, understanding virus-host interaction is important for antiviral drug development. Since de novo nucleotide biosynthesis is essentially required for both host cell metabolism and viral replication, specific catalytic enzymes of these pathways have been explored as potential antiviral targets. In this study, we investigated the role of different enzymatic cascades of nucleotide biosynthesis in hepatitis E virus (HEV) replication. By profiling various pharmacological inhibitors of nucleotide biosynthesis, we found that targeting the early steps of the purine biosynthesis pathway led to the enhancement of HEV replication, whereas targeting the later step resulted in potent antiviral activity via the depletion of purine nucleotide. Furthermore, the inhibition of the pyrimidine pathway resulted in potent anti-HEV activity. Interestingly, all of these inhibitors with anti-HEV activity concurrently triggered the induction of antiviral interferon-stimulated genes (ISGs). Although ISGs are commonly induced by interferons via the JAK-STAT pathway, their induction by nucleotide synthesis inhibitors is completely independent of this classical mechanism. In conclusion, this study revealed an unconventional novel mechanism of cross talk between nucleotide biosynthesis pathways and cellular antiviral immunity in constraining HEV infection. Targeting particular enzymes in nucleotide biosynthesis represents a viable option for antiviral drug development against HEV. HEV is the most common cause of acute viral hepatitis worldwide and is also associated with chronic hepatitis, especially in immunocompromised patients. Although often an acute and self-limiting infection in the general population, HEV can cause severe morbidity and mortality in certain patients, a problem compounded by the lack of FDA-approved anti-HEV medication available. In this study, we have investigated the role of the nucleotide synthesis pathway in HEV infection and its potential for antiviral drug development. We show that targeting the later but not the early steps of the purine synthesis pathway exerts strong anti-HEV activity. In particular, IMP dehydrogenase (IMPDH) is the most important anti-HEV target of this cascade. Importantly, the clinically used IMPDH inhibitors, including mycophenolic acid and ribavirin, have potent anti-HEV activity. Furthermore, targeting the pyrimidine synthesis pathway also exerts potent antiviral activity against HEV. Interestingly, antiviral effects of nucleotide synthesis pathway inhibitors appear to depend on the medication-induced transcription of antiviral interferon-stimulated genes. Thus, this study reveals an unconventional novel mechanism as to how nucleotide synthesis pathway inhibitors can counteract HEV replication.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures









Similar articles
-
RIG-I is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production.Hepatology. 2017 Jun;65(6):1823-1839. doi: 10.1002/hep.29105. Epub 2017 May 3. Hepatology. 2017. PMID: 28195391
-
Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus.Gastroenterology. 2014 Jun;146(7):1775-83. doi: 10.1053/j.gastro.2014.02.036. Epub 2014 Feb 26. Gastroenterology. 2014. PMID: 24582714
-
Drug screening identified gemcitabine inhibiting hepatitis E virus by inducing interferon-like response via activation of STAT1 phosphorylation.Antiviral Res. 2020 Dec;184:104967. doi: 10.1016/j.antiviral.2020.104967. Epub 2020 Oct 31. Antiviral Res. 2020. PMID: 33137361
-
Zinc: A Potential Antiviral Against Hepatitis E Virus Infection?DNA Cell Biol. 2018 Jul;37(7):593-599. doi: 10.1089/dna.2018.4175. Epub 2018 Jun 13. DNA Cell Biol. 2018. PMID: 29897788 Review.
-
Three Distinct Reporter Systems of Hepatitis E Virus and Their Utility as Drug Screening Platforms.Viruses. 2023 Sep 23;15(10):1989. doi: 10.3390/v15101989. Viruses. 2023. PMID: 37896767 Free PMC article. Review.
Cited by
-
Recapitulating hepatitis E virus-host interactions and facilitating antiviral drug discovery in human liver-derived organoids.Sci Adv. 2022 Jan 21;8(3):eabj5908. doi: 10.1126/sciadv.abj5908. Epub 2022 Jan 19. Sci Adv. 2022. PMID: 35044825 Free PMC article.
-
M Segment-Based Minigenome System of Severe Fever with Thrombocytopenia Syndrome Virus as a Tool for Antiviral Drug Screening.Viruses. 2021 Jun 3;13(6):1061. doi: 10.3390/v13061061. Viruses. 2021. PMID: 34205062 Free PMC article.
-
Recent insights for the emerging COVID-19: Drug discovery, therapeutic options and vaccine development.Asian J Pharm Sci. 2021 Jan;16(1):4-23. doi: 10.1016/j.ajps.2020.06.001. Epub 2020 Jul 4. Asian J Pharm Sci. 2021. PMID: 32837565 Free PMC article. Review.
-
A Proximity biotinylation assay with a host protein bait reveals multiple factors modulating enterovirus replication.PLoS Pathog. 2022 Oct 28;18(10):e1010906. doi: 10.1371/journal.ppat.1010906. eCollection 2022 Oct. PLoS Pathog. 2022. PMID: 36306280 Free PMC article.
-
Potential Application of Exosomes in Vaccine Development and Delivery.Pharm Res. 2022 Nov;39(11):2635-2671. doi: 10.1007/s11095-021-03143-4. Epub 2022 Jan 13. Pharm Res. 2022. PMID: 35028802 Free PMC article. Review.
References
-
- Kamar N, Garrouste C, Haagsma EB, Garrigue V, Pischke S, Chauvet C, Dumortier J, Cannesson A, Cassuto-Viguier E, Thervet E, Conti F, Lebray P, Dalton HR, Santella R, Kanaan N, Essig M, Mousson C, Radenne S, Roque-Afonso AM, Izopet J, Rostaing L. 2011. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 140:1481–1489. doi:10.1053/j.gastro.2011.02.050. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
