

Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders
- PMID: 26902584
- PMCID: PMC4835965
- DOI: 10.1080/15548627.2015.1121360
Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders
Abstract
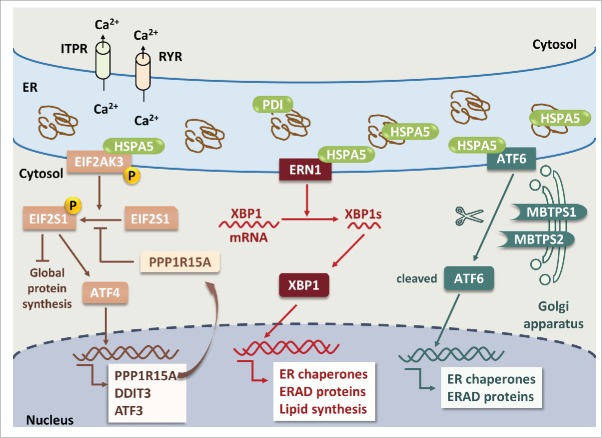
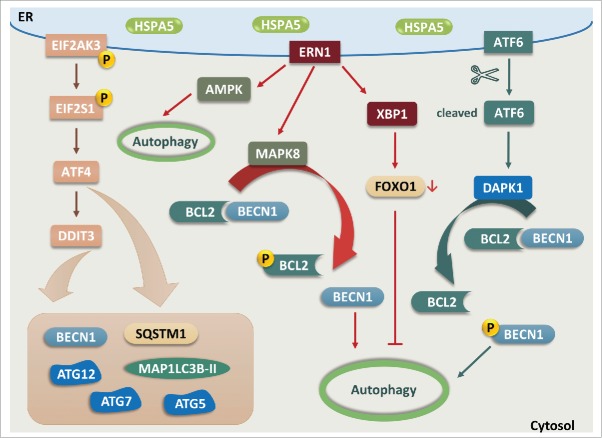
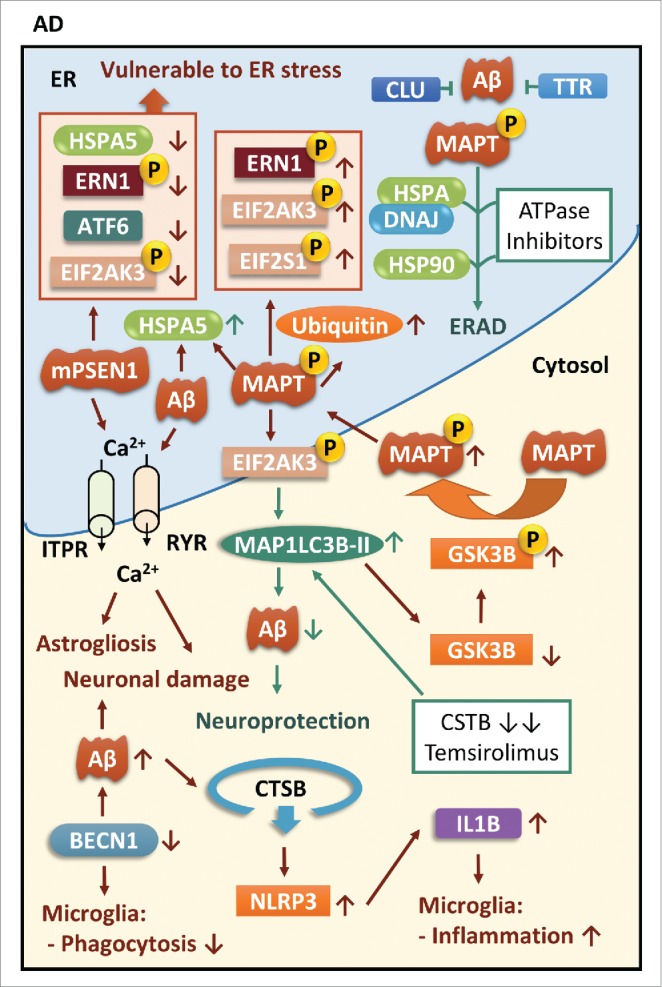
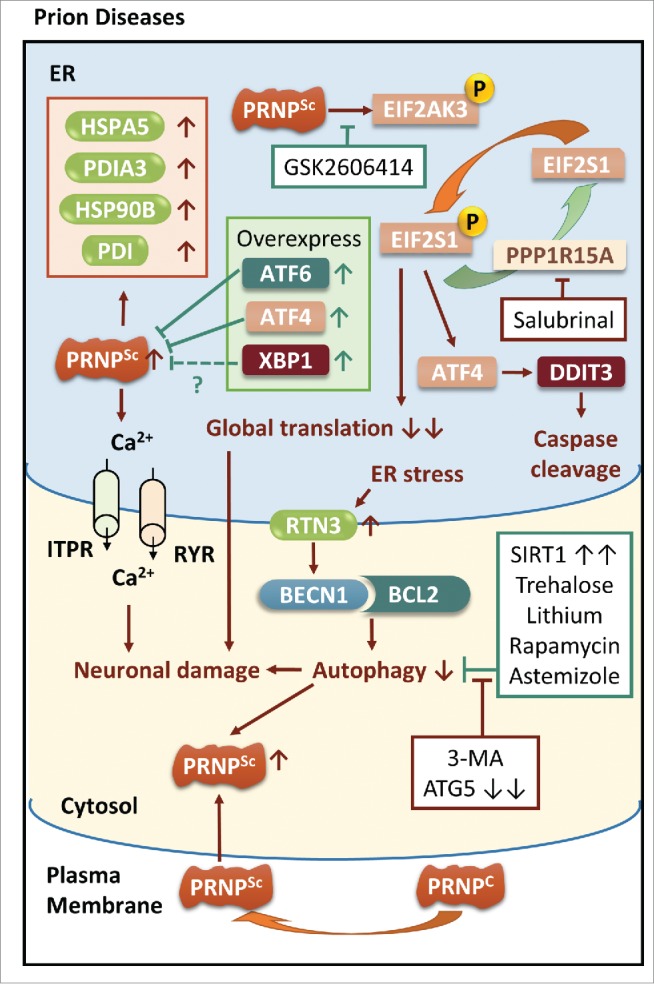
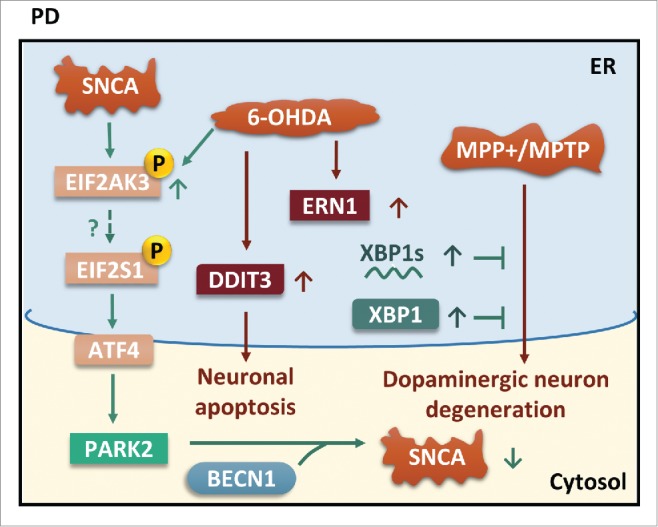
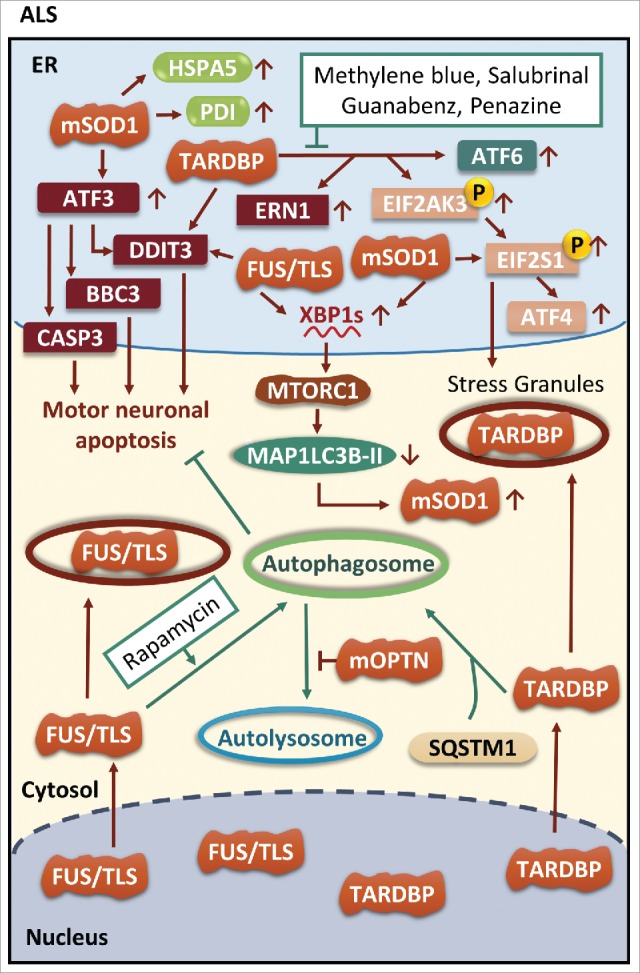
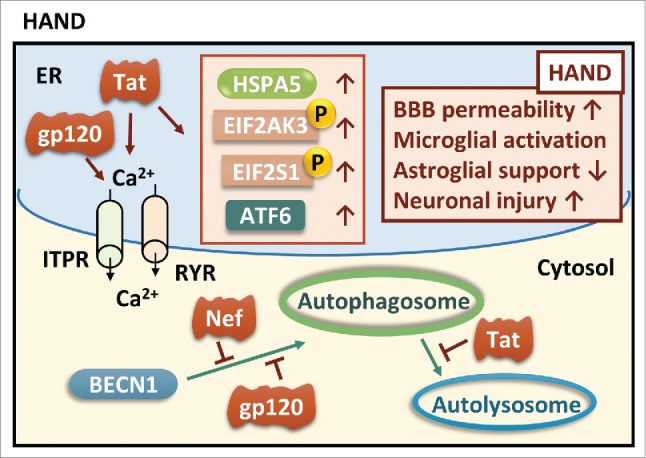
The common underlying feature of most neurodegenerative diseases such as Alzheimer disease (AD), prion diseases, Parkinson disease (PD), and amyotrophic lateral sclerosis (ALS) involves accumulation of misfolded proteins leading to initiation of endoplasmic reticulum (ER) stress and stimulation of the unfolded protein response (UPR). Additionally, ER stress more recently has been implicated in the pathogenesis of HIV-associated neurocognitive disorders (HAND). Autophagy plays an essential role in the clearance of aggregated toxic proteins and degradation of the damaged organelles. There is evidence that autophagy ameliorates ER stress by eliminating accumulated misfolded proteins. Both abnormal UPR and impaired autophagy have been implicated as a causative mechanism in the development of various neurodegenerative diseases. This review highlights recent advances in the field on the role of ER stress and autophagy in AD, prion diseases, PD, ALS and HAND with the involvement of key signaling pathways in these processes and implications for future development of therapeutic strategies.
Keywords: ER stress; Parkinson disease; alzheimer disease; amyotrophic lateral sclerosis and HIV-associated neurocognitive disorders; autophagy; neurodegenerative disorders; prion diseases.
Figures

Similar articles
-
The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress.Int J Mol Sci. 2022 May 24;23(11):5894. doi: 10.3390/ijms23115894. Int J Mol Sci. 2022. PMID: 35682574 Free PMC article. Review.
-
Mechanisms of disordered neurodegenerative function: concepts and facts about the different roles of the protein kinase RNA-like endoplasmic reticulum kinase (PERK).Rev Neurosci. 2018 Jun 27;29(4):387-415. doi: 10.1515/revneuro-2017-0071. Rev Neurosci. 2018. PMID: 29303785 Review.
-
The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration.Curr Mol Med. 2008 May;8(3):157-72. doi: 10.2174/156652408784221324. Curr Mol Med. 2008. PMID: 18473817 Review.
-
The role of endoplasmic reticulum stress in neurodegenerative disease.Apoptosis. 2017 Jan;22(1):1-26. doi: 10.1007/s10495-016-1296-4. Apoptosis. 2017. PMID: 27815720 Review.
-
Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders.J Cell Mol Med. 2011 Oct;15(10):2025-39. doi: 10.1111/j.1582-4934.2011.01374.x. J Cell Mol Med. 2011. PMID: 21722302 Free PMC article. Review.
Cited by
-
The roles and mechanisms of endoplasmic reticulum stress-mediated autophagy in animal viral infections.Vet Res. 2024 Sep 3;55(1):107. doi: 10.1186/s13567-024-01360-4. Vet Res. 2024. PMID: 39227990 Free PMC article. Review.
-
Enhancing calmodulin binding to ryanodine receptor is crucial to limit neuronal cell loss in Alzheimer disease.Sci Rep. 2021 Mar 31;11(1):7289. doi: 10.1038/s41598-021-86822-x. Sci Rep. 2021. PMID: 33790404 Free PMC article.
-
Cinnamaldehyde induces autophagy-mediated cell death through ER stress and epigenetic modification in gastric cancer cells.Acta Pharmacol Sin. 2022 Mar;43(3):712-723. doi: 10.1038/s41401-021-00672-x. Epub 2021 May 12. Acta Pharmacol Sin. 2022. PMID: 33980998 Free PMC article.
-
Neuropharmacologic Approaches to Restore the Brain's Microenvironment.J Neuroimmune Pharmacol. 2016 Sep;11(3):484-94. doi: 10.1007/s11481-016-9686-5. Epub 2016 Jun 28. J Neuroimmune Pharmacol. 2016. PMID: 27352074 Free PMC article. Review.
-
Regulation of morphine-induced synaptic alterations: Role of oxidative stress, ER stress, and autophagy.J Cell Biol. 2016 Oct 24;215(2):245-258. doi: 10.1083/jcb.201605065. Epub 2016 Oct 17. J Cell Biol. 2016. PMID: 27810915 Free PMC article.
References
-
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 2010; 40:238-52; PMID:20965419; http://dx.doi.org/10.1016/j.molcel.2010.10.001 - DOI - PubMed
-
- Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med 2008; 8:157-72; PMID:18473817; http://dx.doi.org/10.2174/156652408784221324 - DOI - PubMed
-
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026 - DOI - PubMed
-
- Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 2012; 44:89-98; PMID:22257884; http://dx.doi.org/10.3858/emm.2012.44.2.031 - DOI - PMC - PubMed
-
- Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, Volsky DJ, Maggirwar SB, Dewhurst S, Masliah E, et al.. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PloS One 2008; 3:e3731; PMID:19009018; http://dx.doi.org/10.1371/journal.pone.0003731 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous