

Determining the Composition and Stability of Protein Complexes Using an Integrated Label-Free and Stable Isotope Labeling Strategy
- PMID: 26867737
- PMCID: PMC4916643
- DOI: 10.1007/978-1-4939-3524-6_3
Determining the Composition and Stability of Protein Complexes Using an Integrated Label-Free and Stable Isotope Labeling Strategy
Abstract
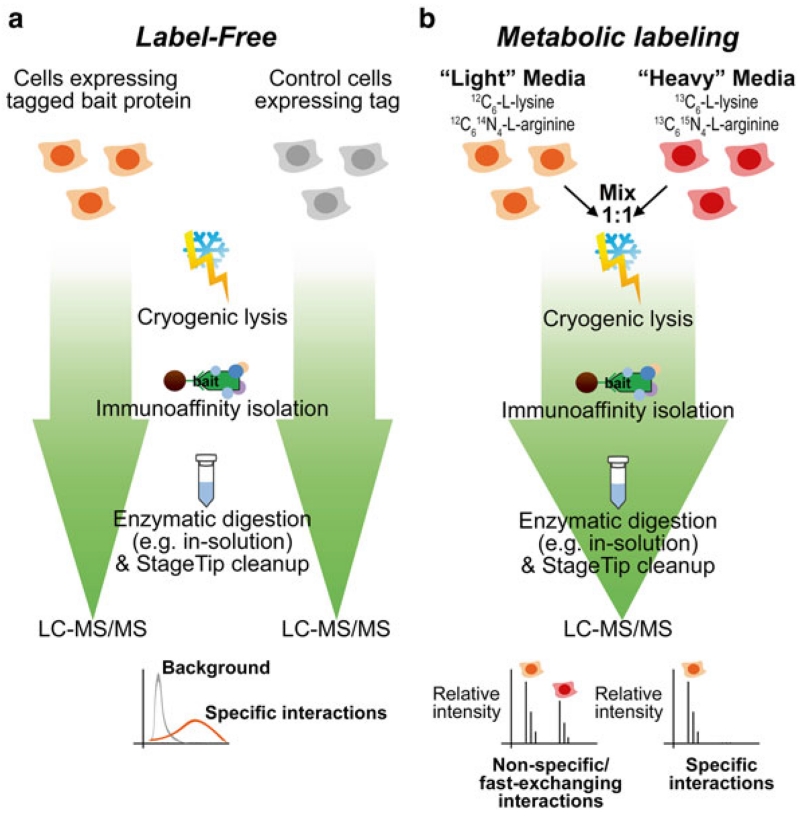
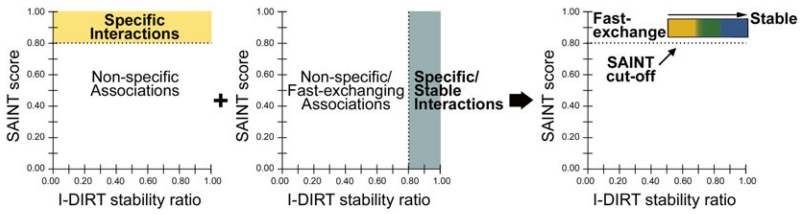
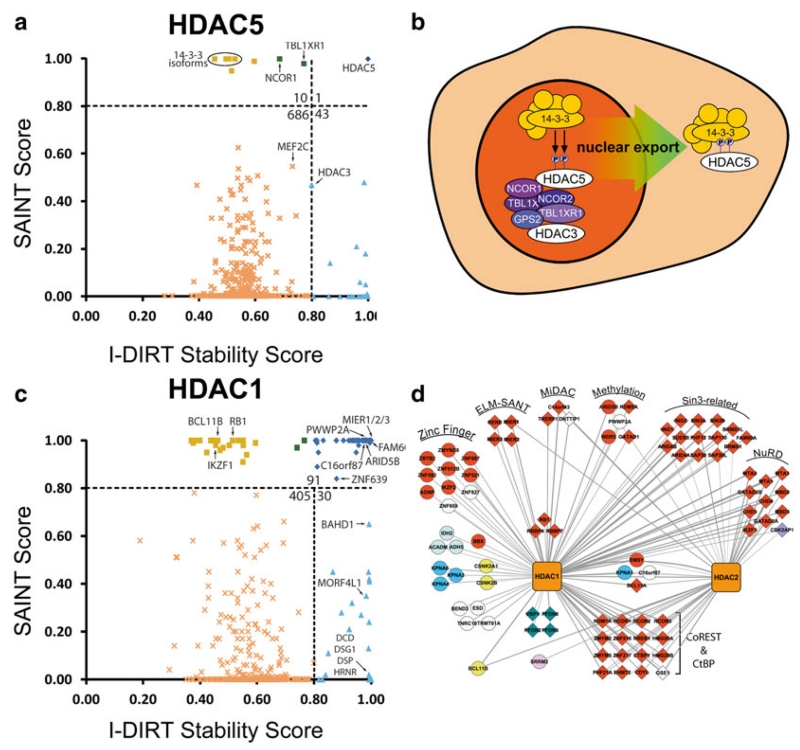
In biological systems, proteins catalyze the fundamental reactions that underlie all cellular functions, including metabolic processes and cell survival and death pathways. These biochemical reactions are rarely accomplished alone. Rather, they involve a concerted effect from many proteins that may operate in a directed signaling pathway and/or may physically associate in a complex to achieve a specific enzymatic activity. Therefore, defining the composition and regulation of protein complexes is critical for understanding cellular functions. In this chapter, we describe an approach that uses quantitative mass spectrometry (MS) to assess the specificity and the relative stability of protein interactions. Isolation of protein complexes from mammalian cells is performed by rapid immunoaffinity purification, and followed by in-solution digestion and high-resolution mass spectrometry analysis. We employ complementary quantitative MS workflows to assess the specificity of protein interactions using label-free MS and statistical analysis, and the relative stability of the interactions using a metabolic labeling technique. For each candidate protein interaction, scores from the two workflows can be correlated to minimize nonspecific background and profile protein complex composition and relative stability.
Keywords: Affinity isolation; I-DIRT; Immunoprecipitation; Label-free quantification; Protein complexes; Protein interactions; SAINT; Stable isotope labeling quantification.
Figures
Similar articles
-
Using stable isotope tagging and mass spectrometry to characterize protein complexes and to detect changes in their composition.Methods Mol Biol. 2007;359:17-35. doi: 10.1007/978-1-59745-255-7_2. Methods Mol Biol. 2007. PMID: 17484108
-
Comparison of stable-isotope labeling with amino acids in cell culture and spectral counting for relative quantification of protein expression.Rapid Commun Mass Spectrom. 2011 Sep 15;25(17):2524-32. doi: 10.1002/rcm.5151. Rapid Commun Mass Spectrom. 2011. PMID: 21818813
-
Resolving protein interactions and complexes by affinity purification followed by label-based quantitative mass spectrometry.Proteomics. 2012 May;12(10):1623-38. doi: 10.1002/pmic.201100438. Proteomics. 2012. PMID: 22610586 Review.
-
Stable isotope labelling methods in mass spectrometry-based quantitative proteomics.J Pharm Biomed Anal. 2015 Sep 10;113:2-20. doi: 10.1016/j.jpba.2015.04.013. Epub 2015 Apr 25. J Pharm Biomed Anal. 2015. PMID: 25956803 Review.
-
Production and use of stable isotope-labeled proteins for absolute quantitative proteomics.Methods Mol Biol. 2011;753:93-115. doi: 10.1007/978-1-61779-148-2_7. Methods Mol Biol. 2011. PMID: 21604118
Cited by
-
Charge-Mediated Pyrin Oligomerization Nucleates Antiviral IFI16 Sensing of Herpesvirus DNA.mBio. 2019 Jul 23;10(4):e01428-19. doi: 10.1128/mBio.01428-19. mBio. 2019. PMID: 31337724 Free PMC article.
-
Hdac4 Interactions in Huntington's Disease Viewed Through the Prism of Multiomics.Mol Cell Proteomics. 2019 Aug 9;18(8 suppl 1):S92-S113. doi: 10.1074/mcp.RA118.001253. Epub 2019 Apr 30. Mol Cell Proteomics. 2019. PMID: 31040226 Free PMC article.
-
Human Sirtuin 2 Localization, Transient Interactions, and Impact on the Proteome Point to Its Role in Intracellular Trafficking.Mol Cell Proteomics. 2016 Oct;15(10):3107-3125. doi: 10.1074/mcp.M116.061333. Epub 2016 Aug 8. Mol Cell Proteomics. 2016. PMID: 27503897 Free PMC article.
-
Mitochondria and Peroxisome Remodeling across Cytomegalovirus Infection Time Viewed through the Lens of Inter-ViSTA.Cell Rep. 2020 Jul 28;32(4):107943. doi: 10.1016/j.celrep.2020.107943. Cell Rep. 2020. PMID: 32726614 Free PMC article.
-
Initiating Events in Direct Cardiomyocyte Reprogramming.Cell Rep. 2018 Feb 13;22(7):1913-1922. doi: 10.1016/j.celrep.2018.01.047. Cell Rep. 2018. PMID: 29444441 Free PMC article.
References
-
- Rigaut G, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17(10):1030–1032. - PubMed
-
- Cristea IM, et al. Fluorescent proteins as proteomic probes. Mol Cell Proteomics. 2005;4(12):1933–1941. - PubMed
-
- Sueda S, Tanaka H, Yamagishi M. A biotin-based protein tagging system. Anal Biochem. 2009;393(2):189–195. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
