

The Genomes of Three Uneven Siblings: Footprints of the Lifestyles of Three Trichoderma Species
- PMID: 26864432
- PMCID: PMC4771370
- DOI: 10.1128/MMBR.00040-15
The Genomes of Three Uneven Siblings: Footprints of the Lifestyles of Three Trichoderma Species
Abstract
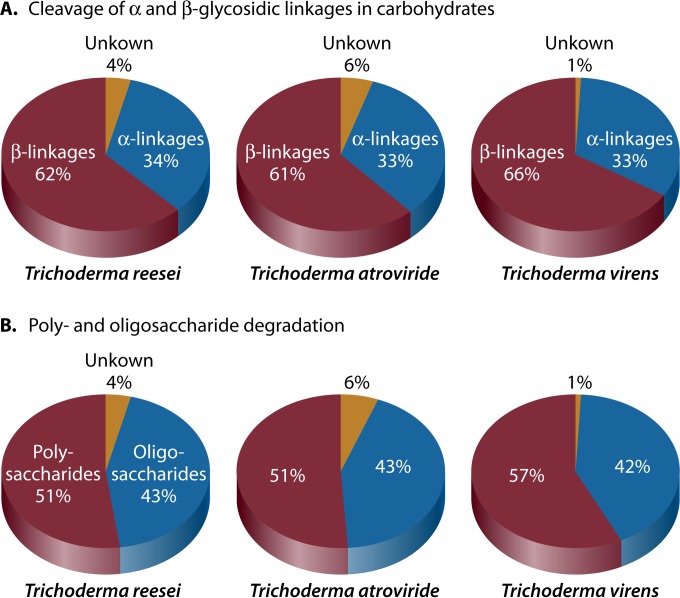
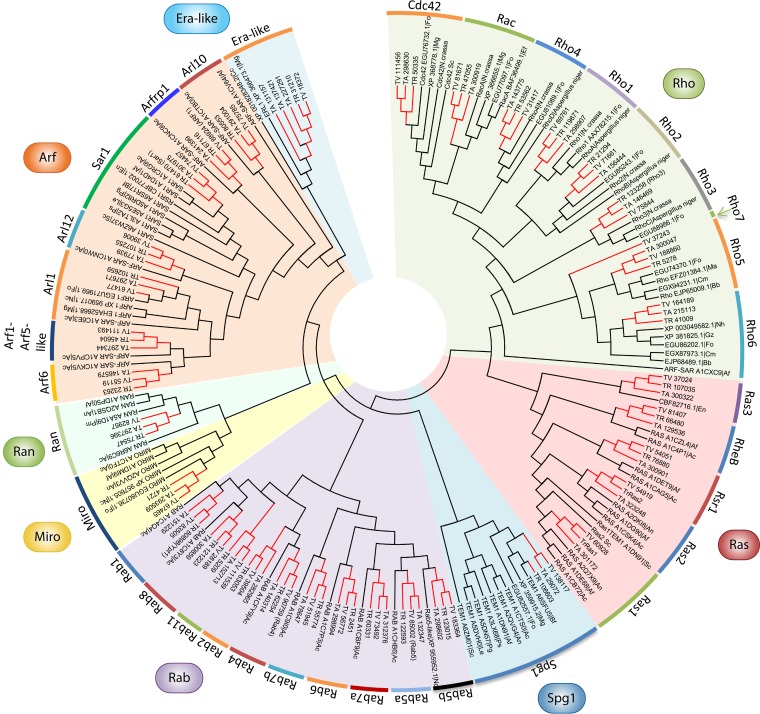
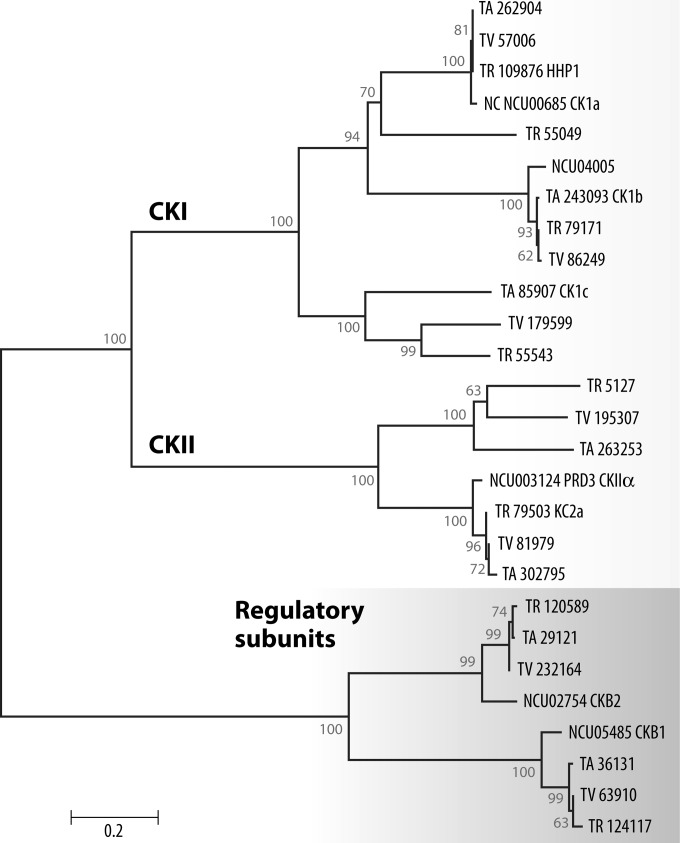
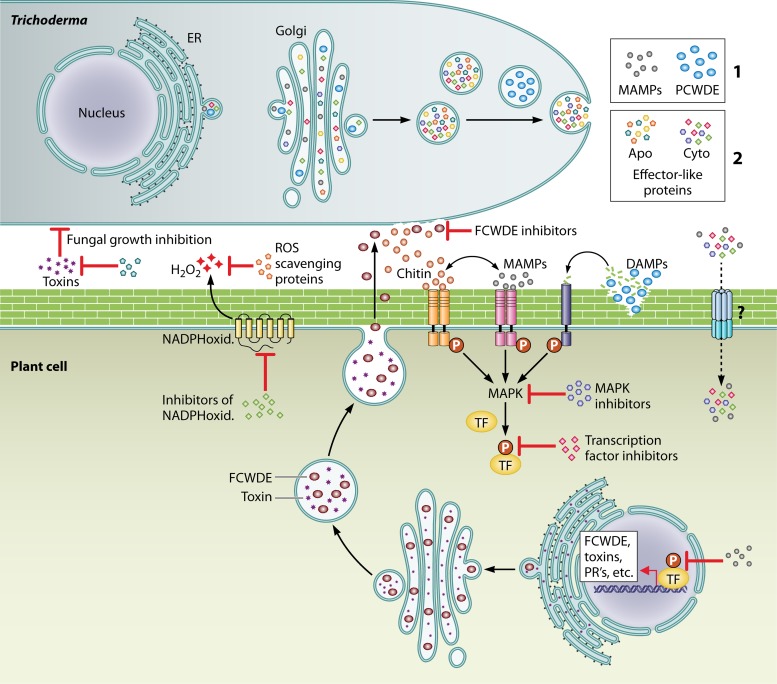
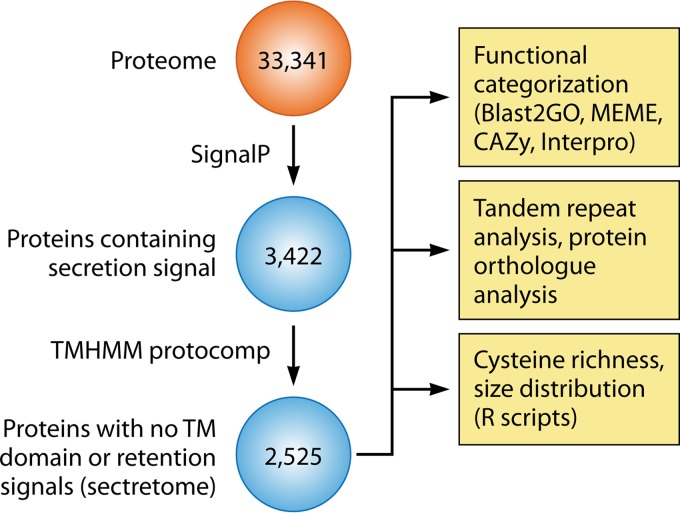
The genus Trichoderma contains fungi with high relevance for humans, with applications in enzyme production for plant cell wall degradation and use in biocontrol. Here, we provide a broad, comprehensive overview of the genomic content of these species for "hot topic" research aspects, including CAZymes, transport, transcription factors, and development, along with a detailed analysis and annotation of less-studied topics, such as signal transduction, genome integrity, chromatin, photobiology, or lipid, sulfur, and nitrogen metabolism in T. reesei, T. atroviride, and T. virens, and we open up new perspectives to those topics discussed previously. In total, we covered more than 2,000 of the predicted 9,000 to 11,000 genes of each Trichoderma species discussed, which is >20% of the respective gene content. Additionally, we considered available transcriptome data for the annotated genes. Highlights of our analyses include overall carbohydrate cleavage preferences due to the different genomic contents and regulation of the respective genes. We found light regulation of many sulfur metabolic genes. Additionally, a new Golgi 1,2-mannosidase likely involved in N-linked glycosylation was detected, as were indications for the ability of Trichoderma spp. to generate hybrid galactose-containing N-linked glycans. The genomic inventory of effector proteins revealed numerous compounds unique to Trichoderma, and these warrant further investigation. We found interesting expansions in the Trichoderma genus in several signaling pathways, such as G-protein-coupled receptors, RAS GTPases, and casein kinases. A particularly interesting feature absolutely unique to T. atroviride is the duplication of the alternative sulfur amino acid synthesis pathway.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures


Similar articles
-
Complete Genome Sequences and Genome-Wide Characterization of Trichoderma Biocontrol Agents Provide New Insights into their Evolution and Variation in Genome Organization, Sexual Development, and Fungal-Plant Interactions.Microbiol Spectr. 2021 Dec 22;9(3):e0066321. doi: 10.1128/Spectrum.00663-21. Epub 2021 Dec 15. Microbiol Spectr. 2021. PMID: 34908505 Free PMC article.
-
Comparative analysis of the repertoire of G protein-coupled receptors of three species of the fungal genus Trichoderma.BMC Microbiol. 2013 May 16;13:108. doi: 10.1186/1471-2180-13-108. BMC Microbiol. 2013. PMID: 23679152 Free PMC article.
-
Phylogenomic analysis of polyketide synthase-encoding genes in Trichoderma.Microbiology (Reading). 2012 Jan;158(Pt 1):147-154. doi: 10.1099/mic.0.053462-0. Epub 2011 Nov 17. Microbiology (Reading). 2012. PMID: 22096146
-
Biology and biotechnology of Trichoderma.Appl Microbiol Biotechnol. 2010 Jul;87(3):787-99. doi: 10.1007/s00253-010-2632-1. Epub 2010 May 12. Appl Microbiol Biotechnol. 2010. PMID: 20461510 Free PMC article. Review.
-
Novel traits of Trichoderma predicted through the analysis of its secretome.FEMS Microbiol Lett. 2012 Dec;337(1):1-9. doi: 10.1111/j.1574-6968.2012.02665.x. Epub 2012 Sep 18. FEMS Microbiol Lett. 2012. PMID: 22924408 Free PMC article. Review.
Cited by
-
Protein phosphatases regulate growth, development, cellulases and secondary metabolism in Trichoderma reesei.Sci Rep. 2019 Jul 29;9(1):10995. doi: 10.1038/s41598-019-47421-z. Sci Rep. 2019. PMID: 31358805 Free PMC article.
-
Histidine kinase two-component response regulators Ssk1, Skn7 and Rim15 differentially control growth, developmental and volatile organic compounds emissions as stress responses in Trichoderma atroviride.Curr Res Microb Sci. 2022 May 18;3:100139. doi: 10.1016/j.crmicr.2022.100139. eCollection 2022. Curr Res Microb Sci. 2022. PMID: 35909598 Free PMC article.
-
The Kinase USK1 Regulates Cellulase Gene Expression and Secondary Metabolite Biosynthesis in Trichoderma reesei.Front Microbiol. 2020 May 20;11:974. doi: 10.3389/fmicb.2020.00974. eCollection 2020. Front Microbiol. 2020. PMID: 32508786 Free PMC article.
-
Integrated Translatome and Proteome: Approach for Accurate Portraying of Widespread Multifunctional Aspects of Trichoderma.Front Microbiol. 2017 Aug 29;8:1602. doi: 10.3389/fmicb.2017.01602. eCollection 2017. Front Microbiol. 2017. PMID: 28900417 Free PMC article. Review.
-
Omics Analyses of Trichoderma reesei CBS999.97 and QM6a Indicate the Relevance of Female Fertility to Carbohydrate-Active Enzyme and Transporter Levels.Appl Environ Microbiol. 2017 Oct 31;83(22):e01578-17. doi: 10.1128/AEM.01578-17. Print 2017 Nov 15. Appl Environ Microbiol. 2017. PMID: 28916559 Free PMC article.
References
-
- Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy MG. 2014. Biotechnology and biology of Trichoderma. Elsevier, Oxford, United Kingdom.
-
- Mukherjee PK, Horwitz BA, Singh US, Mukherjee M, Schmoll M. 2013. Trichoderma: biology and applications. CAB International, Oxfordshire, United Kingdom.
-
- Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, Mukherjee M, Kredics L, Alcaraz LD, Aerts A, Antal Z, Atanasova L, Cervantes-Badillo MG, Challacombe J, Chertkov O, McCluskey K, Coulpier F, Deshpande N, von Dohren H, Ebbole DJ, Esquivel-Naranjo EU, Fekete E, Flipphi M, Glaser F, Gomez-Rodriguez EY, Gruber S, Han C, Henrissat B, Hermosa R, Hernandez-Onate M, Karaffa L, Kosti I, Le Crom S, Lindquist E, Lucas S, Lubeck M, Lubeck PS, Margeot A, Metz B, Misra M, Nevalainen H, Omann M, Packer N, Perrone G, Uresti-Rivera EE, Salamov A, Schmoll M, Seiboth B, Shapiro H, Sukno S, Tamayo-Ramos JA, Tisch D, Wiest A, Wilkinson HH, Zhang M, Coutinho PM, Kenerley CM, Monte E, Baker SE, Grigoriev IV. 2011. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12:R40. doi:10.1186/gb-2011-12-4-r40. - DOI - PMC - PubMed
-
- Schmoll M, Seiboth B, Druzhinina I, Kubicek CP. 2014. Genomics analysis of biocontrol species and industrial enzyme producers from the genus Trichoderma, p 233–266. In Esser K, Nowrousian M (ed), The Mycota XIII. Springer, Berlin, Germany.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
