

Spiked proteomic standard dataset for testing label-free quantitative software and statistical methods
- PMID: 26862574
- PMCID: PMC4706616
- DOI: 10.1016/j.dib.2015.11.063
Spiked proteomic standard dataset for testing label-free quantitative software and statistical methods
Abstract
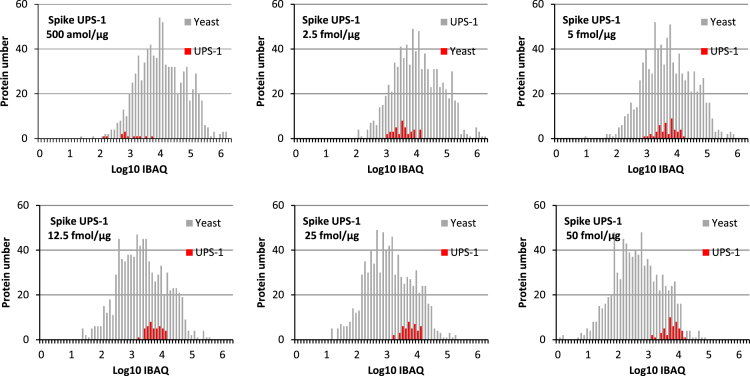
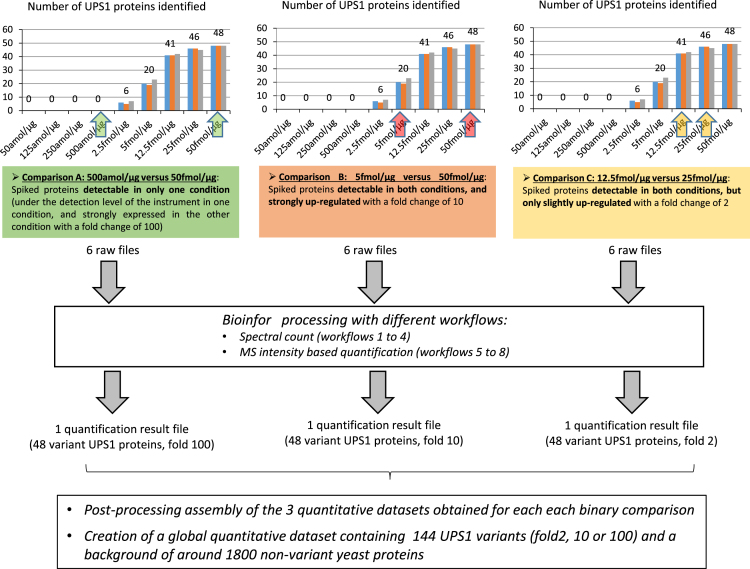
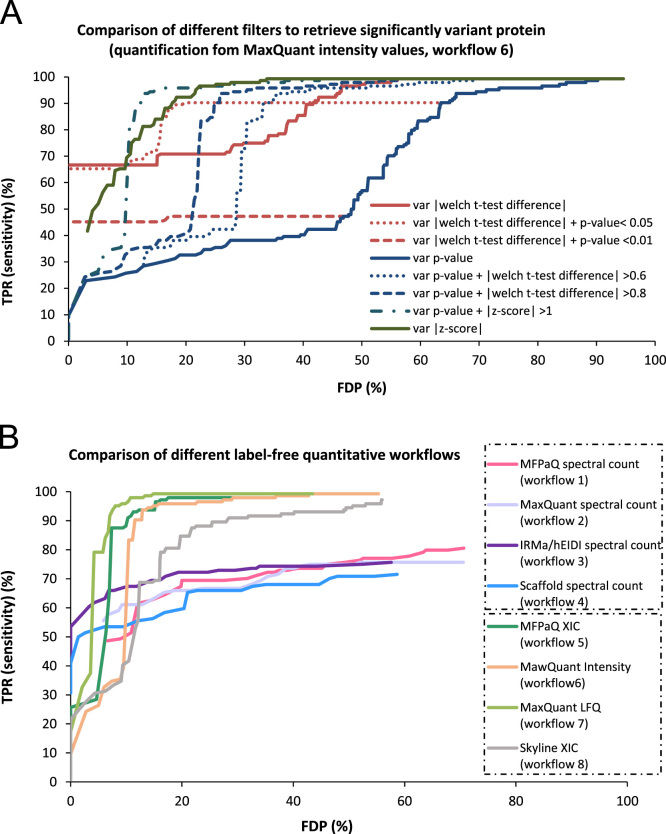
This data article describes a controlled, spiked proteomic dataset for which the "ground truth" of variant proteins is known. It is based on the LC-MS analysis of samples composed of a fixed background of yeast lysate and different spiked amounts of the UPS1 mixture of 48 recombinant proteins. It can be used to objectively evaluate bioinformatic pipelines for label-free quantitative analysis, and their ability to detect variant proteins with good sensitivity and low false discovery rate in large-scale proteomic studies. More specifically, it can be useful for tuning software tools parameters, but also testing new algorithms for label-free quantitative analysis, or for evaluation of downstream statistical methods. The raw MS files can be downloaded from ProteomeXchange with identifier PXD001819. Starting from some raw files of this dataset, we also provide here some processed data obtained through various bioinformatics tools (including MaxQuant, Skyline, MFPaQ, IRMa-hEIDI and Scaffold) in different workflows, to exemplify the use of such data in the context of software benchmarking, as discussed in details in the accompanying manuscript [1]. The experimental design used here for data processing takes advantage of the different spike levels introduced in the samples composing the dataset, and processed data are merged in a single file to facilitate the evaluation and illustration of software tools results for the detection of variant proteins with different absolute expression levels and fold change values.
Figures
Similar articles
-
Benchmarking quantitative label-free LC-MS data processing workflows using a complex spiked proteomic standard dataset.J Proteomics. 2016 Jan 30;132:51-62. doi: 10.1016/j.jprot.2015.11.011. Epub 2015 Nov 14. J Proteomics. 2016. PMID: 26585461
-
DIA proteomics data from a UPS1-spiked E.coli protein mixture processed with six software tools.Data Brief. 2022 Jan 31;41:107829. doi: 10.1016/j.dib.2022.107829. eCollection 2022 Apr. Data Brief. 2022. PMID: 35198661 Free PMC article.
-
Extensive and Accurate Benchmarking of DIA Acquisition Methods and Software Tools Using a Complex Proteomic Standard.J Proteome Res. 2021 Oct 1;20(10):4801-4814. doi: 10.1021/acs.jproteome.1c00490. Epub 2021 Sep 2. J Proteome Res. 2021. PMID: 34472865
-
Dataset containing physiological amounts of spike-in proteins into murine C2C12 background as a ground truth quantitative LC-MS/MS reference.Data Brief. 2022 Jul 4;43:108435. doi: 10.1016/j.dib.2022.108435. eCollection 2022 Aug. Data Brief. 2022. PMID: 35845101 Free PMC article.
-
Mass spectrometry data from identification of host-defense related proteins using label-free quantitative proteomic analysis of milk whey from cows with Staphylococcus aureus subclinical mastitis.Data Brief. 2019 Jan 10;22:909-913. doi: 10.1016/j.dib.2019.01.013. eCollection 2019 Feb. Data Brief. 2019. PMID: 30733978 Free PMC article.
Cited by
-
Robust determination of differential abundance in shotgun proteomics using nonparametric statistics.Mol Omics. 2018 Dec 3;14(6):424-436. doi: 10.1039/c8mo00077h. Mol Omics. 2018. PMID: 30259924 Free PMC article.
-
A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation.Brief Bioinform. 2018 Nov 27;19(6):1344-1355. doi: 10.1093/bib/bbx054. Brief Bioinform. 2018. PMID: 28575146 Free PMC article.
-
IceR improves proteome coverage and data completeness in global and single-cell proteomics.Nat Commun. 2021 Aug 9;12(1):4787. doi: 10.1038/s41467-021-25077-6. Nat Commun. 2021. PMID: 34373457 Free PMC article.
-
Methods and Challenges for Computational Data Analysis for DNA Adductomics.Chem Res Toxicol. 2019 Nov 18;32(11):2156-2168. doi: 10.1021/acs.chemrestox.9b00196. Epub 2019 Nov 6. Chem Res Toxicol. 2019. PMID: 31549505 Free PMC article.
-
FlexStat: combinatory differentially expressed protein extraction.Bioinform Adv. 2024 Apr 11;4(1):vbae056. doi: 10.1093/bioadv/vbae056. eCollection 2024. Bioinform Adv. 2024. PMID: 38681522 Free PMC article.
References
-
- Ramus C., Hovasse A., Marcellin M., Hesse A.M., Mouton-Barbosa E., Bouyssié D. Benchmarking quantitative label-free LC-MS data processing workflows using a complex spiked proteomic standard dataset. J. Proteom. 2016;132:51–62. - PubMed
-
- Bouyssie D., Gonzalez de Peredo A., Mouton E., Albigot R., Roussel L., Ortega N. Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelial cells. Mol. Cell. Proteom.: MCP. 2007;6:1621–1637. - PubMed
-
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. - PubMed
-
- Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J.V. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009;4:698–705. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
