

Feedback activation of neurofibromin terminates growth factor-induced Ras activation
- PMID: 26861207
- PMCID: PMC4746934
- DOI: 10.1186/s12964-016-0128-z
Feedback activation of neurofibromin terminates growth factor-induced Ras activation
Abstract
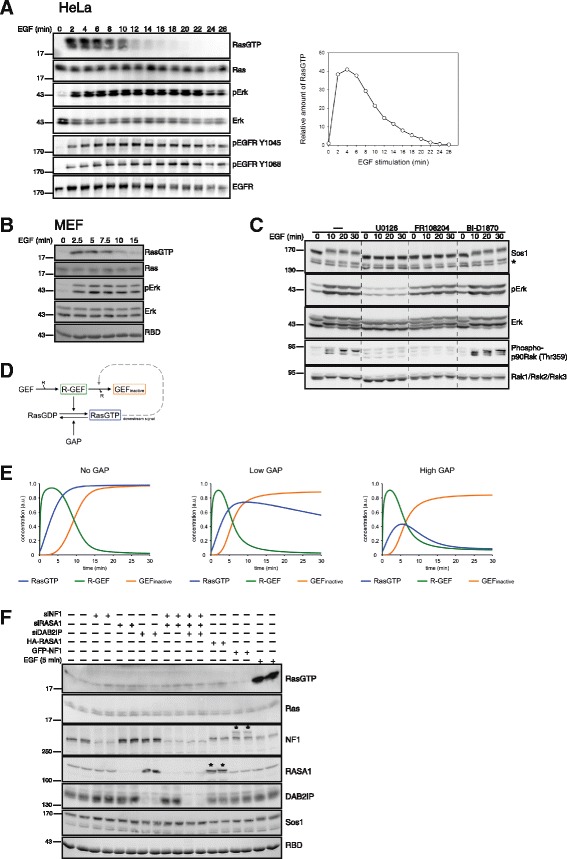
Background: Growth factors induce a characteristically short-lived Ras activation in cells emerging from quiescence. Extensive work has shown that transient as opposed to sustained Ras activation is critical for the induction of mitogenic programs. Mitogen-induced accumulation of active Ras-GTP results from increased nucleotide exchange driven by the nucleotide exchange factor Sos. In contrast, the mechanism accounting for signal termination and prompt restoration of basal Ras-GTP levels is unclear, but has been inferred to involve feedback inhibition of Sos. Remarkably, how GTP-hydrolase activating proteins (GAPs) participate in controlling the rise and fall of Ras-GTP levels is unknown.
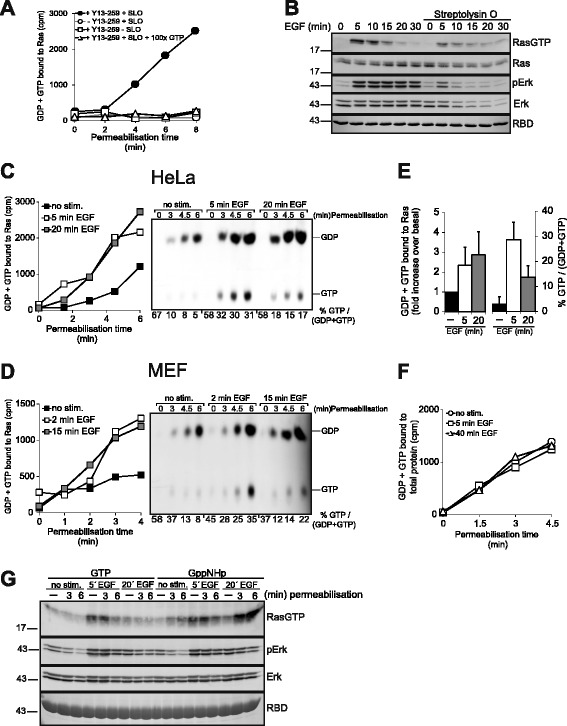
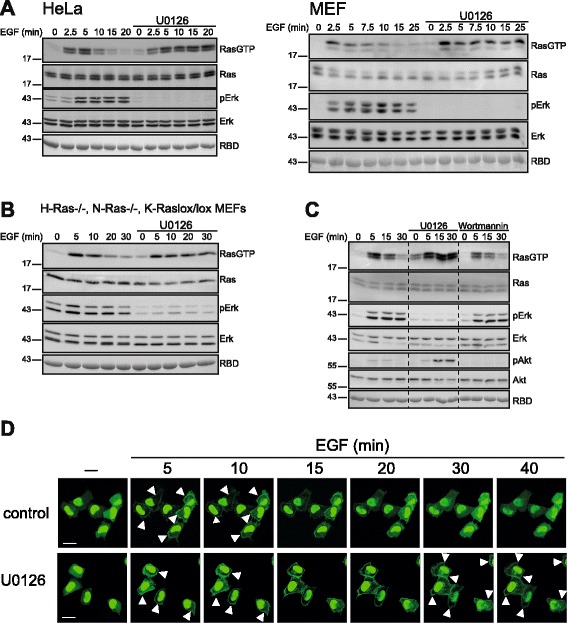
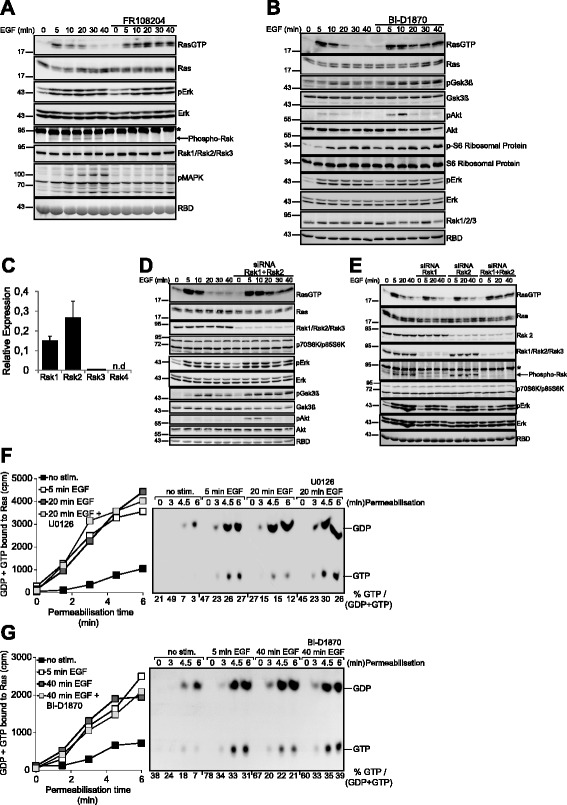
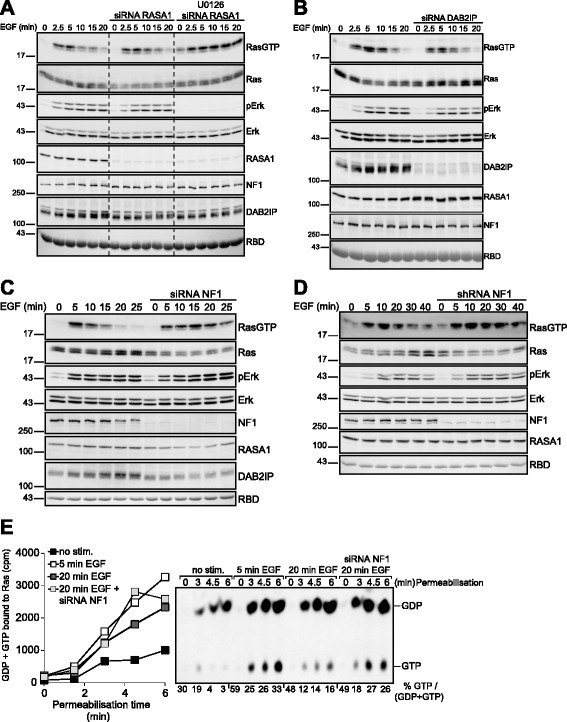
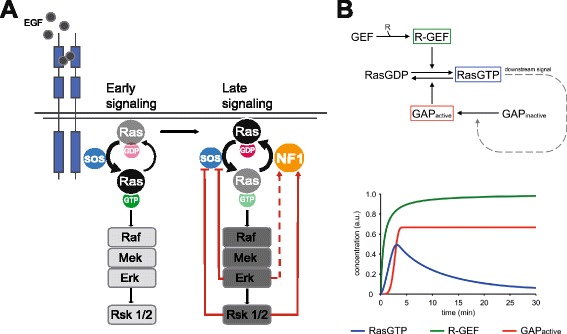
Results: Monitoring nucleotide exchange of Ras in permeabilized cells we find, unexpectedly, that the decline of growth factor-induced Ras-GTP levels proceeds in the presence of unabated high nucleotide exchange, pointing to GAP activation as a major mechanism of signal termination. Experiments with non-hydrolysable GTP analogues and mathematical modeling confirmed and rationalized the presence of high GAP activity as Ras-GTP levels decline in a background of high nucleotide exchange. Using pharmacological and genetic approaches we document a raised activity of the neurofibromatosis type I tumor suppressor Ras-GAP neurofibromin and an involvement of Rsk1 and Rsk2 in the down-regulation of Ras-GTP levels.
Conclusions: Our findings show that, in addition to feedback inhibition of Sos, feedback stimulation of the RasGAP neurofibromin enforces termination of the Ras signal in the context of growth-factor signaling. These findings ascribe a precise role to neurofibromin in growth factor-dependent control of Ras activity and illustrate how, by engaging Ras-GAP activity, mitogen-challenged cells play safe to ensure a timely termination of the Ras signal irrespectively of the reigning rate of nucleotide exchange.
Figures
Similar articles
-
Ras activation in response to phorbol ester proceeds independently of the EGFR via an unconventional nucleotide-exchange factor system in COS-7 cells.Biochem J. 2006 Sep 1;398(2):243-56. doi: 10.1042/BJ20060160. Biochem J. 2006. PMID: 16709153 Free PMC article.
-
Aberrant Ras regulation and reduced p190 tyrosine phosphorylation in cells lacking p120-Gap.Mol Cell Biol. 1997 Apr;17(4):1840-7. doi: 10.1128/MCB.17.4.1840. Mol Cell Biol. 1997. PMID: 9121432 Free PMC article.
-
Mechanism of SHIP-mediated inhibition of insulin- and platelet-derived growth factor-stimulated mitogen-activated protein kinase activity in 3T3-L1 adipocytes.Mol Endocrinol. 2005 Feb;19(2):421-30. doi: 10.1210/me.2004-0096. Epub 2004 Oct 14. Mol Endocrinol. 2005. PMID: 15486046
-
Ras activation revisited: role of GEF and GAP systems.Biol Chem. 2015 Aug;396(8):831-48. doi: 10.1515/hsz-2014-0257. Biol Chem. 2015. PMID: 25781681 Review.
-
Control of ras activation.Cancer Surv. 1996;27:87-100. Cancer Surv. 1996. PMID: 8909796 Review.
Cited by
-
ERK1/2-RSK2 Signaling in Regulation of ERα-Mediated Responses.Endocrinology. 2022 Sep 1;163(9):bqac106. doi: 10.1210/endocr/bqac106. Endocrinology. 2022. PMID: 35880639 Free PMC article. Review.
-
RSK1 promotes mammalian axon regeneration by inducing the synthesis of regeneration-related proteins.PLoS Biol. 2022 Jun 1;20(6):e3001653. doi: 10.1371/journal.pbio.3001653. eCollection 2022 Jun. PLoS Biol. 2022. PMID: 35648763 Free PMC article.
-
New insights into RAS biology reinvigorate interest in mathematical modeling of RAS signaling.Semin Cancer Biol. 2019 Feb;54:162-173. doi: 10.1016/j.semcancer.2018.02.008. Epub 2018 Mar 5. Semin Cancer Biol. 2019. PMID: 29518522 Free PMC article. Review.
-
Active GTPase Pulldown Protocol.Methods Mol Biol. 2021;2262:117-135. doi: 10.1007/978-1-0716-1190-6_7. Methods Mol Biol. 2021. PMID: 33977474
-
Therapeutic targeting of p90 ribosomal S6 kinase.Front Cell Dev Biol. 2023 Dec 19;11:1297292. doi: 10.3389/fcell.2023.1297292. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 38169775 Free PMC article. Review.
References
-
- Muroya K, Hattori S, Nakamura S. Nerve growth factor induces rapid accumulation of the GTP-bound form of p21ras in rat pheochromocytoma PC12 cells. Oncogene. 1992;7:277–81. - PubMed
-
- Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288(Pt 2):351–5. doi: 10.1042/bj2880351. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
