

SeqTools: visual tools for manual analysis of sequence alignments
- PMID: 26801397
- PMCID: PMC4724122
- DOI: 10.1186/s13104-016-1847-3
SeqTools: visual tools for manual analysis of sequence alignments
Abstract
Background: Manual annotation is essential to create high-quality reference alignments and annotation. Annotators need to be able to view sequence alignments in detail. The SeqTools package provides three tools for viewing different types of sequence alignment: Blixem is a many-to-one browser of pairwise alignments, displaying multiple match sequences aligned against a single reference sequence; Dotter provides a graphical dot-plot view of a single pairwise alignment; and Belvu is a multiple sequence alignment viewer, editor, and phylogenetic tool. These tools were originally part of the AceDB genome database system but have been completely rewritten to make them generally available as a standalone package of greatly improved function.
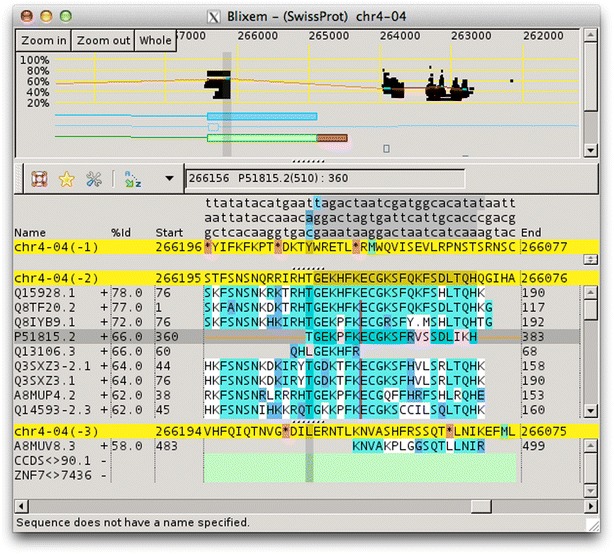
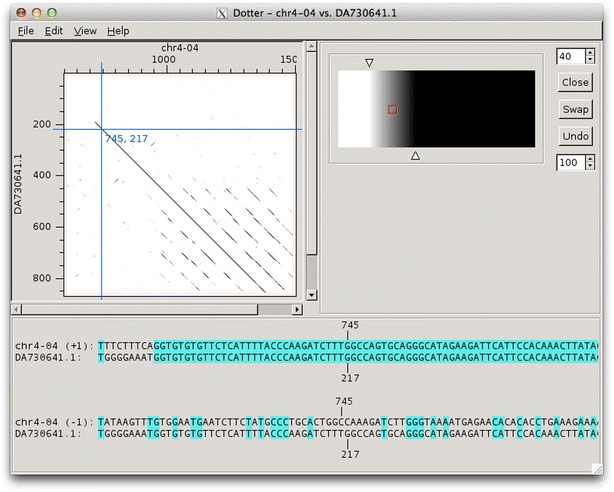
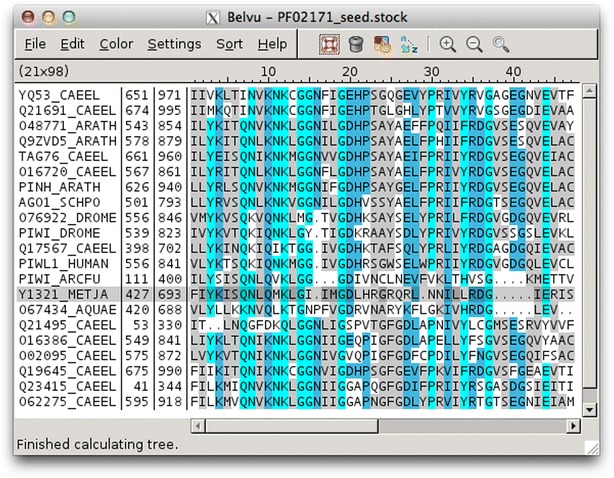
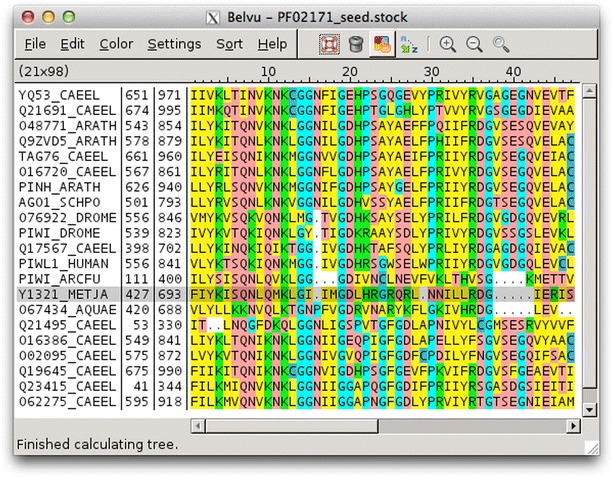
Findings: Blixem is used by annotators to give a detailed view of the evidence for particular gene models. Blixem displays the gene model positions and the match sequences aligned against the genomic reference sequence. Annotators use this for many reasons, including to check the quality of an alignment, to find missing/misaligned sequence and to identify splice sites and polyA sites and signals. Dotter is used to give a dot-plot representation of a particular pairwise alignment. This is used to identify sequence that is not represented (or is misrepresented) and to quickly compare annotated gene models with transcriptional and protein evidence that putatively supports them. Belvu is used to analyse conservation patterns in multiple sequence alignments and to perform a combination of manual and automatic processing of the alignment. High-quality reference alignments are essential if they are to be used as a starting point for further automatic alignment generation.
Conclusions: While there are many different alignment tools available, the SeqTools package provides unique functionality that annotators have found to be essential for analysing sequence alignments as part of the manual annotation process.
Figures

Similar articles
-
GATA: a graphic alignment tool for comparative sequence analysis.BMC Bioinformatics. 2005 Jan 17;6:9. doi: 10.1186/1471-2105-6-9. BMC Bioinformatics. 2005. PMID: 15655071 Free PMC article.
-
Combo: a whole genome comparative browser.Bioinformatics. 2006 Jul 15;22(14):1782-3. doi: 10.1093/bioinformatics/btl193. Epub 2006 May 18. Bioinformatics. 2006. PMID: 16709588
-
CGAT: a comparative genome analysis tool for visualizing alignments in the analysis of complex evolutionary changes between closely related genomes.BMC Bioinformatics. 2006 Oct 24;7:472. doi: 10.1186/1471-2105-7-472. BMC Bioinformatics. 2006. PMID: 17062155 Free PMC article.
-
Multiple sequence alignment: in pursuit of homologous DNA positions.Genome Res. 2007 Feb;17(2):127-35. doi: 10.1101/gr.5232407. Genome Res. 2007. PMID: 17272647 Review.
-
Comparative Genome Annotation.Methods Mol Biol. 2024;2802:165-187. doi: 10.1007/978-1-0716-3838-5_7. Methods Mol Biol. 2024. PMID: 38819560 Review.
Cited by
-
Characterization of defense responses against bacterial pathogens in duckweeds lacking EDS1.New Phytol. 2022 Dec;236(5):1838-1855. doi: 10.1111/nph.18453. Epub 2022 Sep 25. New Phytol. 2022. PMID: 36052715 Free PMC article.
-
Refining wet lab experiments with in silico searches: A rational quest for diagnostic peptides in visceral leishmaniasis.PLoS Negl Trop Dis. 2019 May 6;13(5):e0007353. doi: 10.1371/journal.pntd.0007353. eCollection 2019 May. PLoS Negl Trop Dis. 2019. PMID: 31059497 Free PMC article.
-
Deep learning insights into the architecture of the mammalian egg-sperm fusion synapse.Elife. 2024 Apr 26;13:RP93131. doi: 10.7554/eLife.93131. Elife. 2024. PMID: 38666763 Free PMC article.
-
S-plot2: Rapid Visual and Statistical Analysis of Genomic Sequences.Evol Bioinform Online. 2018 Sep 17;14:1176934318797354. doi: 10.1177/1176934318797354. eCollection 2018. Evol Bioinform Online. 2018. PMID: 30245567 Free PMC article.
-
In silico discovery of the myxosortases that process MYXO-CTERM and three novel prokaryotic C-terminal protein-sorting signals that share invariant Cys residues.J Bacteriol. 2024 Jan 25;206(1):e0017323. doi: 10.1128/jb.00173-23. Epub 2023 Dec 12. J Bacteriol. 2024. PMID: 38084967 Free PMC article.
References
-
- Durbin R, Thierry-Mieg J. The ACeDB genome database. In: Suhai S, editor. Computational methods in genome research. Berlin: Springer; 1994. p. 45–55. doi:10.1007/978-1-4615-2451-9_4.
-
- Sonnhammer ELL, Durbin R.A workbench for large-scale sequence homology analysis. Comput Appl Biosci (CABIOS). 1994;10(3):301–7. (1994). doi:10.1093/bioinformatics/10.3.301. http://bioinformatics.oxfordjournals.org/content/10/3/301.full.pdf+html. - PubMed
-
- Sonnhammer ELL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic dna and protein sequence analysis. Comput Appl Biosci (CABIOS). 1995;167(1–2):1–10. doi:10.1016/0378-1119(95)00714-8. http://bioinformatics.oxfordjournals.org/content/10/3/301.full.pdf+html. - PubMed
-
- Harrow J, Denoeud F, Frankish A, Reymond A, Chen C-K, Chrast J, Lagarde J, Gilbert J, Storey R, Swarbreck D, Rossier C, Ucla C, Hubbard T, Antonarakis S, Guigo R. Gencode: producing a reference annotation for encode. Genome Biol. 2006;7(Suppl. 1):4. doi: 10.1186/gb-2006-7-s1-s4. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
