

Reduced Glucose Sensation Can Increase the Fitness of Saccharomyces cerevisiae Lacking Mitochondrial DNA
- PMID: 26751567
- PMCID: PMC4709096
- DOI: 10.1371/journal.pone.0146511
Reduced Glucose Sensation Can Increase the Fitness of Saccharomyces cerevisiae Lacking Mitochondrial DNA
Abstract
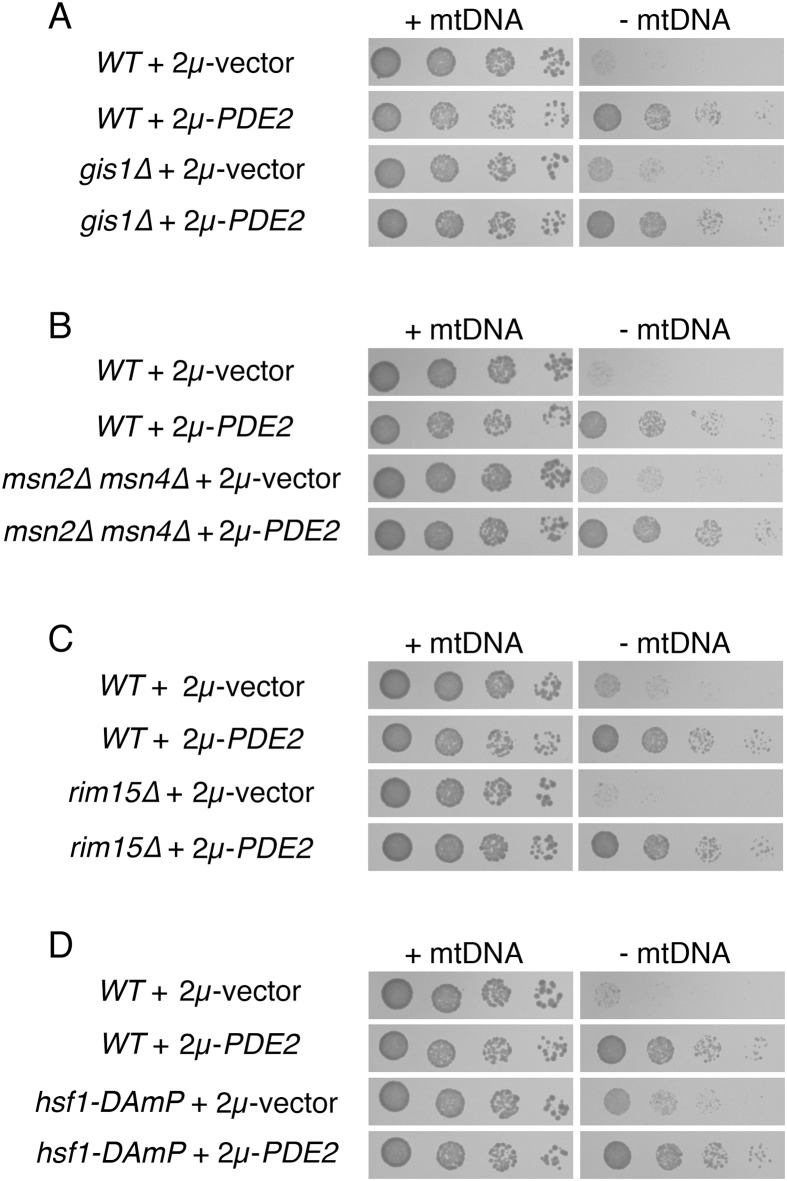
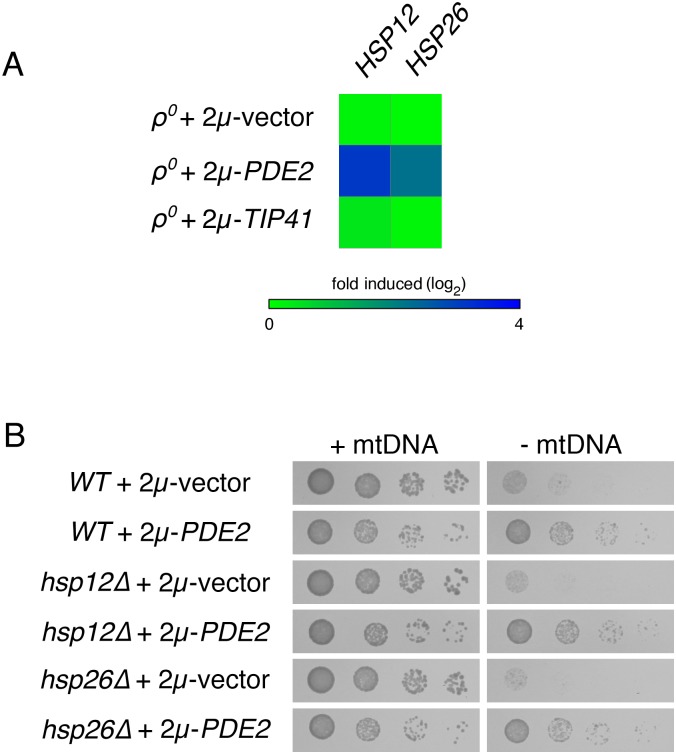
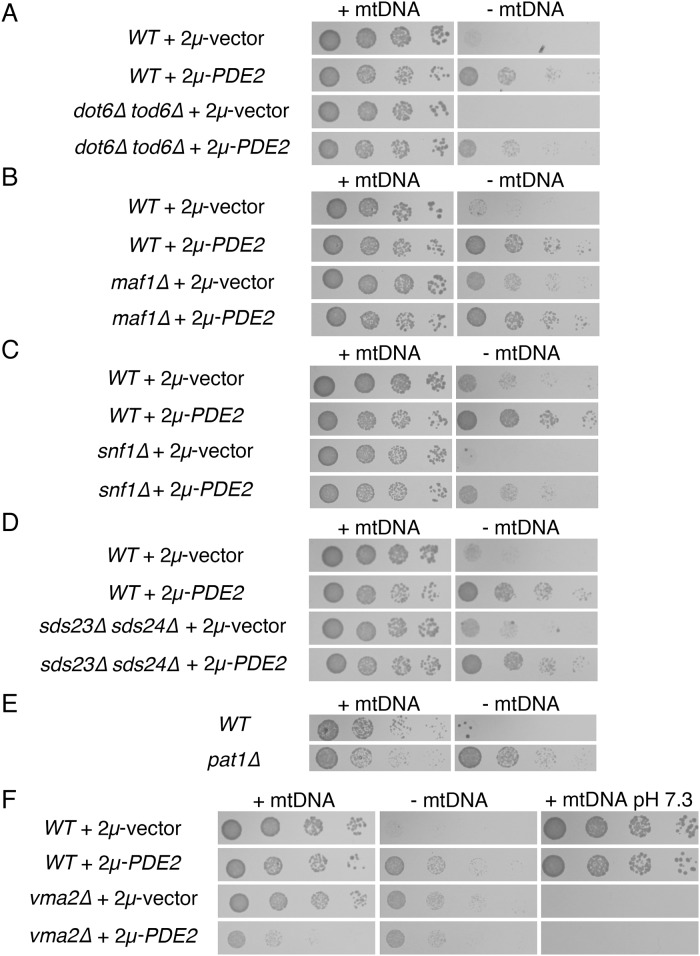
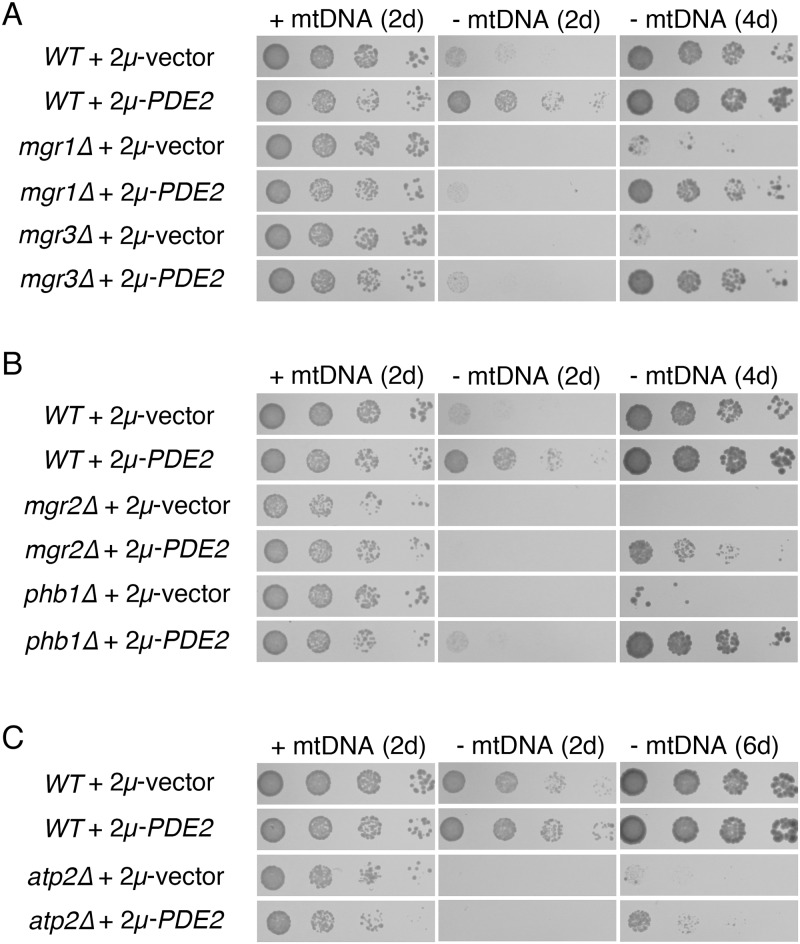
Damage to the mitochondrial genome (mtDNA) can lead to diseases for which there are no clearly effective treatments. Since mitochondrial function and biogenesis are controlled by the nutrient environment of the cell, it is possible that perturbation of conserved, nutrient-sensing pathways may successfully treat mitochondrial disease. We found that restricting glucose or otherwise reducing the activity of the protein kinase A (PKA) pathway can lead to improved proliferation of Saccharomyces cerevisiae cells lacking mtDNA and that the transcriptional response to mtDNA loss is reduced in cells with diminished PKA activity. We have excluded many pathways and proteins from being individually responsible for the benefits provided to cells lacking mtDNA by PKA inhibition, and we found that robust import of mitochondrial polytopic membrane proteins may be required in order for cells without mtDNA to receive the full benefits of PKA reduction. Finally, we have discovered that the transcription of genes involved in arginine biosynthesis and aromatic amino acid catabolism is altered after mtDNA damage. Our results highlight the potential importance of nutrient detection and availability on the outcome of mitochondrial dysfunction.
Conflict of interest statement
Figures
Similar articles
-
Deletion of conserved protein phosphatases reverses defects associated with mitochondrial DNA damage in Saccharomyces cerevisiae.Proc Natl Acad Sci U S A. 2014 Jan 28;111(4):1473-8. doi: 10.1073/pnas.1312399111. Epub 2014 Jan 13. Proc Natl Acad Sci U S A. 2014. PMID: 24474773 Free PMC article.
-
Regulation of mitochondrial protein import by cytosolic kinases.Cell. 2011 Jan 21;144(2):227-39. doi: 10.1016/j.cell.2010.12.015. Epub 2011 Jan 6. Cell. 2011. PMID: 21215441
-
The conserved translocase Tim17 prevents mitochondrial DNA loss.Hum Mol Genet. 2009 Jan 1;18(1):65-74. doi: 10.1093/hmg/ddn313. Epub 2008 Sep 30. Hum Mol Genet. 2009. PMID: 18826960 Free PMC article.
-
Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae.Mol Microbiol. 1999 Sep;33(5):904-18. doi: 10.1046/j.1365-2958.1999.01538.x. Mol Microbiol. 1999. PMID: 10476026 Review.
-
Folding and biogenesis of mitochondrial small Tim proteins.Int J Mol Sci. 2013 Aug 13;14(8):16685-705. doi: 10.3390/ijms140816685. Int J Mol Sci. 2013. PMID: 23945562 Free PMC article. Review.
Cited by
-
Gene dosage adaptations to mtDNA depletion and mitochondrial protein stress in budding yeast.G3 (Bethesda). 2024 Feb 7;14(2):jkad272. doi: 10.1093/g3journal/jkad272. G3 (Bethesda). 2024. PMID: 38126114 Free PMC article.
-
Loss of major nutrient sensing and signaling pathways suppresses starvation lethality in electron transport chain mutants.Mol Biol Cell. 2021 Dec 1;32(22):ar39. doi: 10.1091/mbc.E21-06-0314. Epub 2021 Oct 20. Mol Biol Cell. 2021. PMID: 34668730 Free PMC article.
-
mPOS is a novel mitochondrial trigger of cell death - implications for neurodegeneration.FEBS Lett. 2018 Mar;592(5):759-775. doi: 10.1002/1873-3468.12894. Epub 2017 Nov 14. FEBS Lett. 2018. PMID: 29090463 Free PMC article. Review.
-
Pkh1p-Ypk1p and Pkh1p-Sch9p Pathways Are Activated by Acetic Acid to Induce a Mitochondrial-Dependent Regulated Cell Death.Oxid Med Cell Longev. 2020 Apr 2;2020:7095078. doi: 10.1155/2020/7095078. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 32318242 Free PMC article.
-
Transcriptomic and fatty acid analyses of Neochloris aquatica grown under different nitrogen concentration.Funct Integr Genomics. 2022 Jun;22(3):407-421. doi: 10.1007/s10142-022-00838-8. Epub 2022 Mar 14. Funct Integr Genomics. 2022. PMID: 35286570
References
-
- Voet D, Voet JG. Biochemistry, 4th Edition 4 ed. Wiley Global Education; 2010.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
