

Solution NMR Spectroscopy for the Study of Enzyme Allostery
- PMID: 26734986
- PMCID: PMC4937494
- DOI: 10.1021/acs.chemrev.5b00541
Solution NMR Spectroscopy for the Study of Enzyme Allostery
Abstract
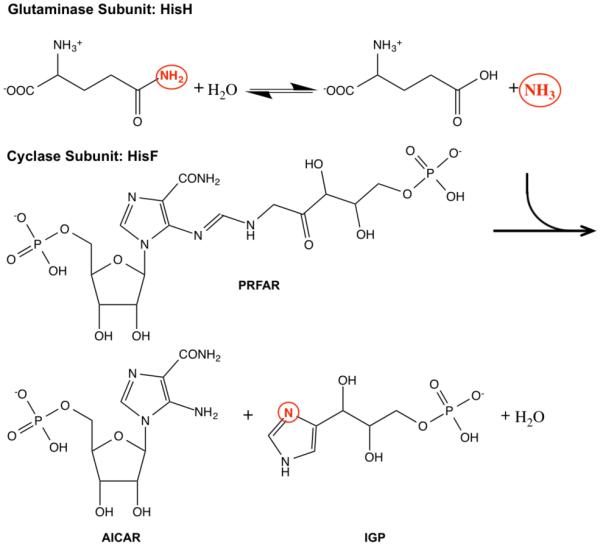
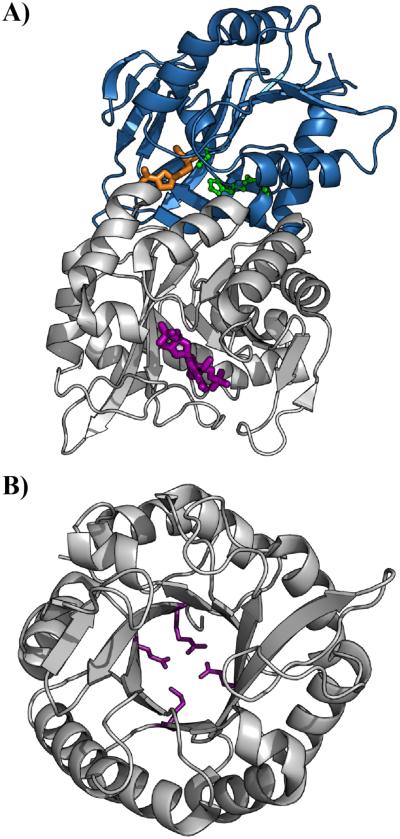
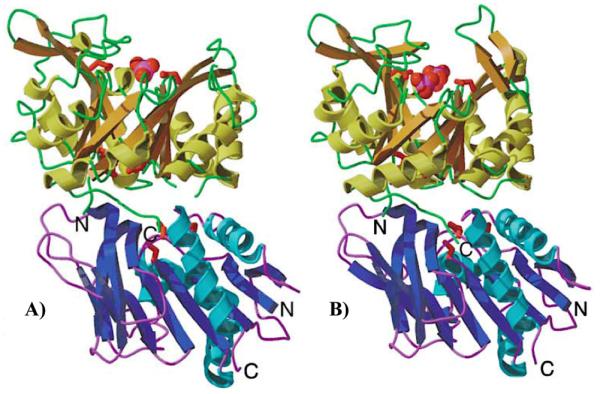
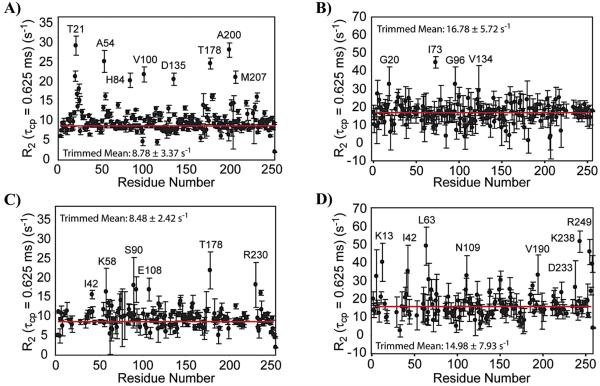
Allostery is a ubiquitous biological regulatory process in which distant binding sites within a protein or enzyme are functionally and thermodynamically coupled. Allosteric interactions play essential roles in many enzymological mechanisms, often facilitating formation of enzyme-substrate complexes and/or product release. Thus, elucidating the forces that drive allostery is critical to understanding the complex transformations of biomolecules. Currently, a number of models exist to describe allosteric behavior, taking into account energetics as well as conformational rearrangements and fluctuations. In the following Review, we discuss the use of solution NMR techniques designed to probe allosteric mechanisms in enzymes. NMR spectroscopy is unequaled in its ability to detect structural and dynamical changes in biomolecules, and the case studies presented herein demonstrate the range of insights to be gained from this valuable method. We also provide a detailed technical discussion of several specialized NMR experiments that are ideally suited for the study of enzymatic allostery.
Figures
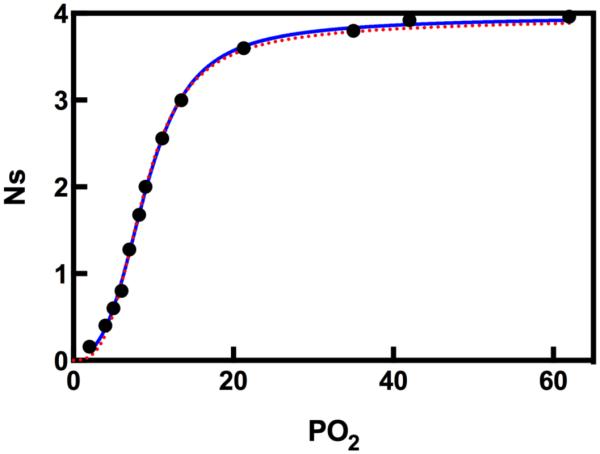
 ) and KNF (—) models in describing O2 binding to Hb. The data points are taken from Monod, Wyman, Changeaux. The curves are non-linear least squares fits with Equations (1) and (2).
) and KNF (—) models in describing O2 binding to Hb. The data points are taken from Monod, Wyman, Changeaux. The curves are non-linear least squares fits with Equations (1) and (2).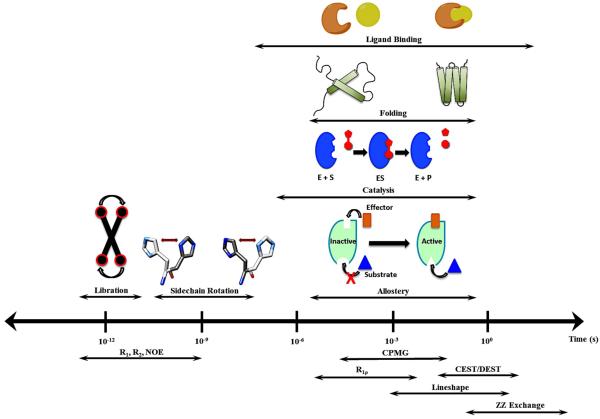
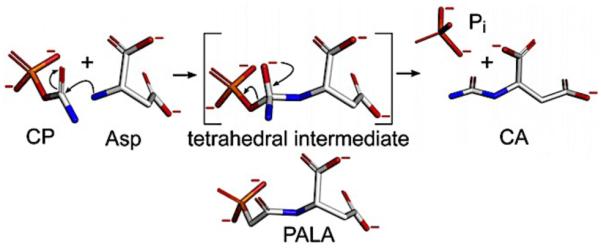
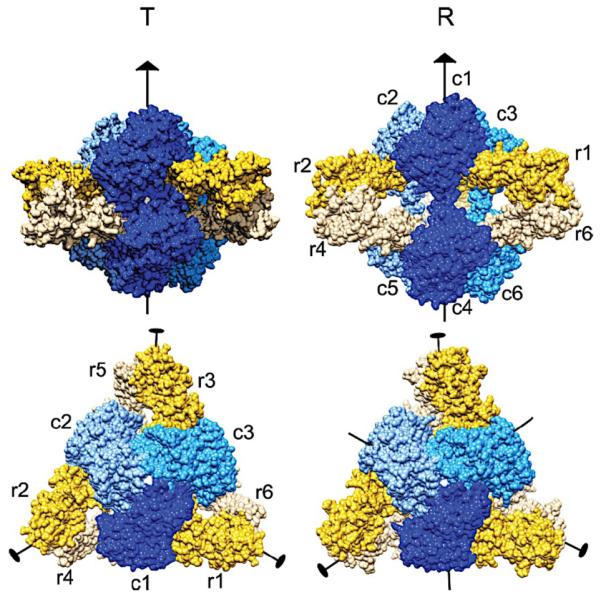

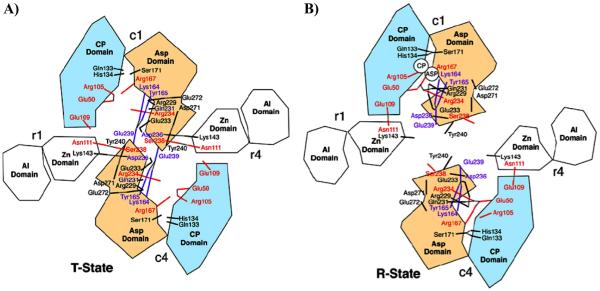
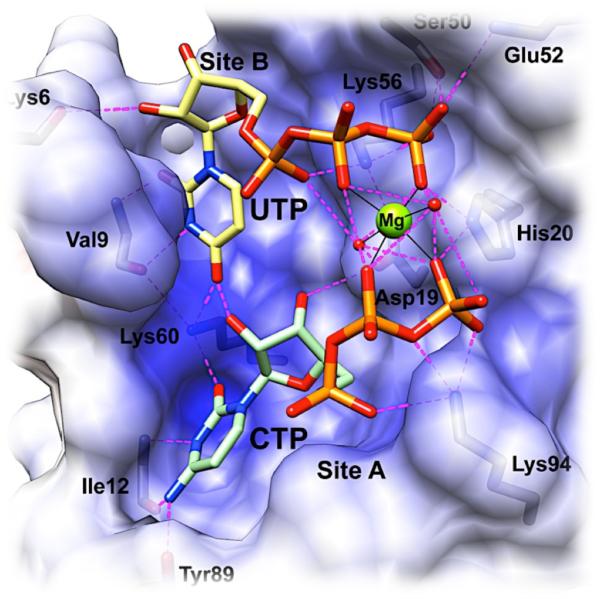




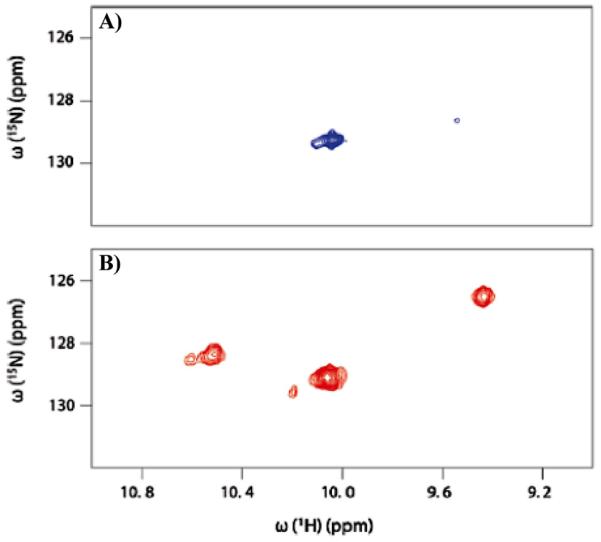
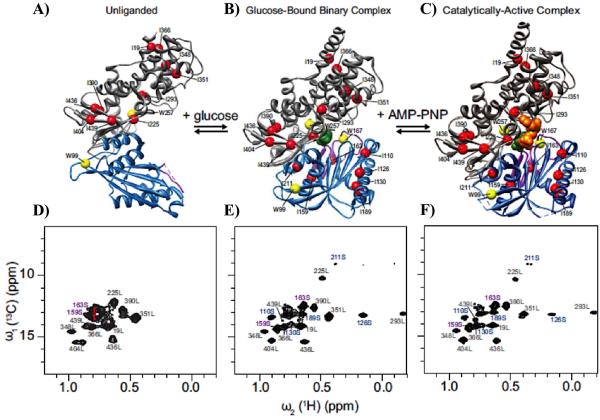
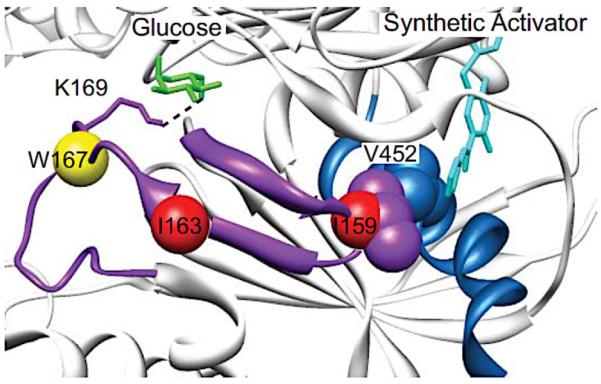
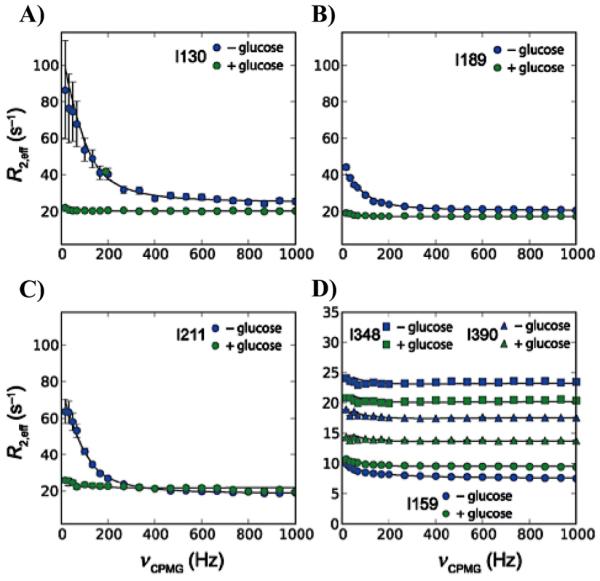
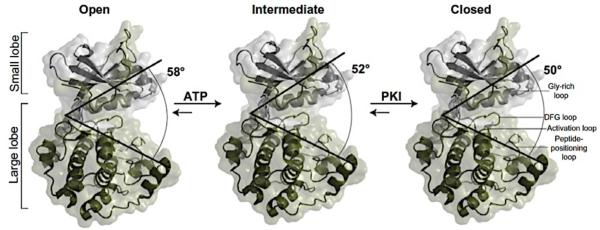
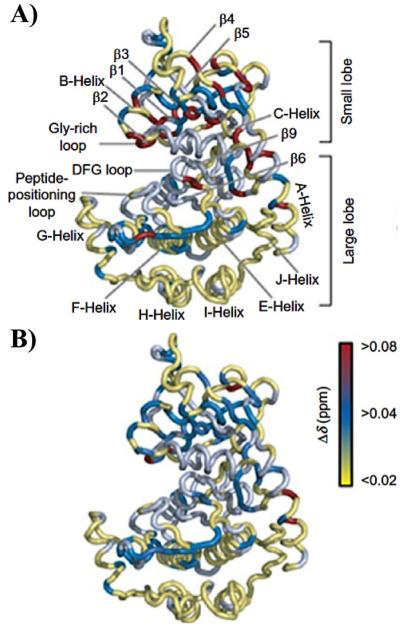
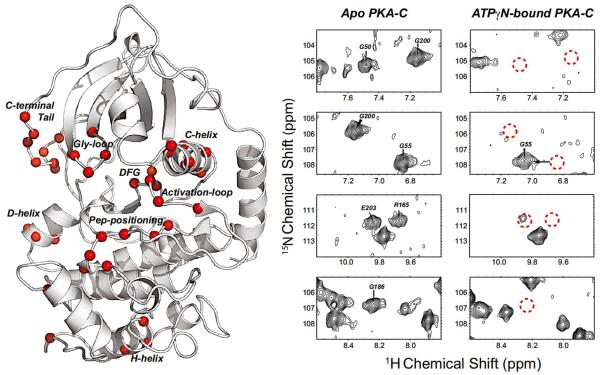
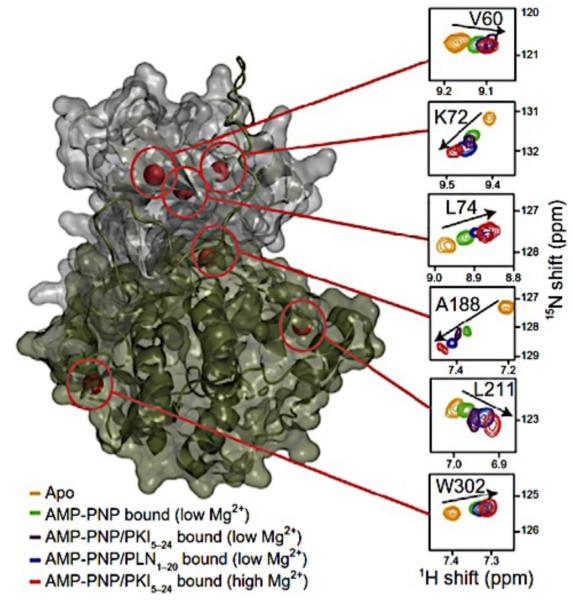
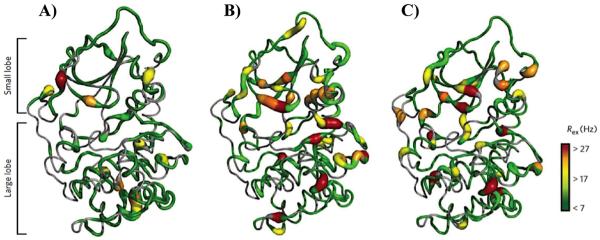
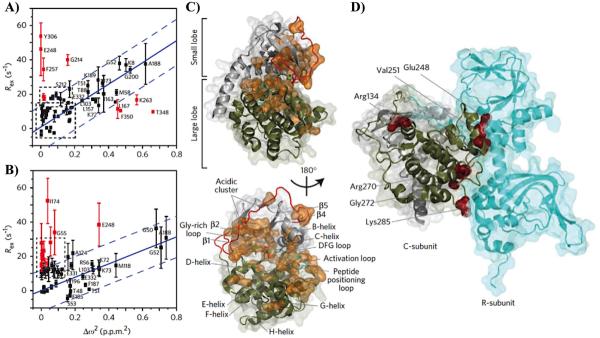
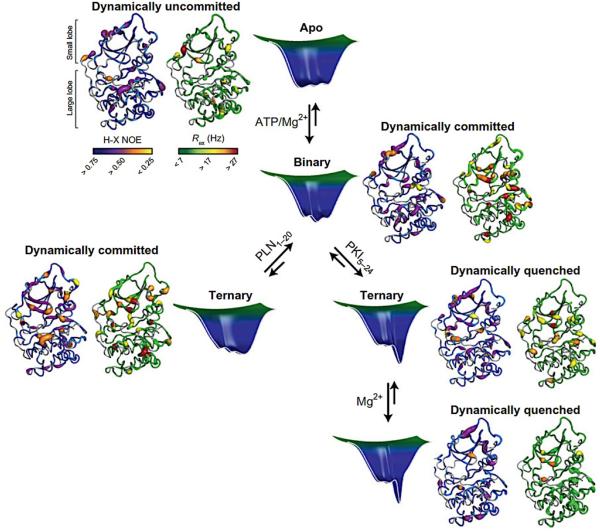
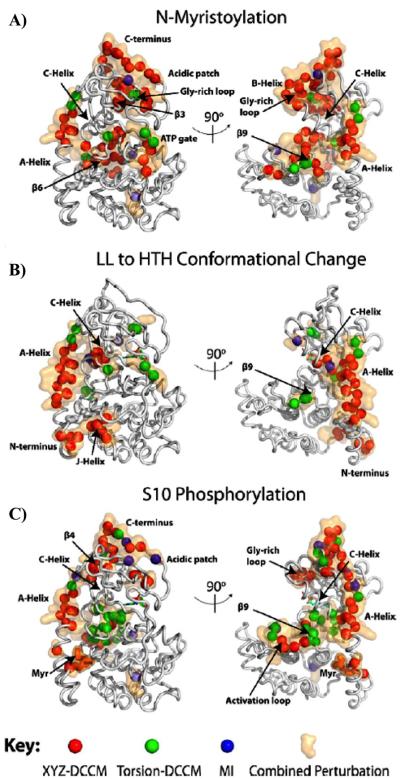
Similar articles
-
NMR and computational methods for molecular resolution of allosteric pathways in enzyme complexes.Biophys Rev. 2020 Feb;12(1):155-174. doi: 10.1007/s12551-019-00609-z. Epub 2019 Dec 14. Biophys Rev. 2020. PMID: 31838649 Free PMC article. Review.
-
NMR tools to detect protein allostery.Curr Opin Struct Biol. 2024 Jun;86:102792. doi: 10.1016/j.sbi.2024.102792. Epub 2024 Mar 1. Curr Opin Struct Biol. 2024. PMID: 38428364 Review.
-
Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function.Prog Nucl Magn Reson Spectrosc. 2016 Feb;92-93:1-17. doi: 10.1016/j.pnmrs.2015.11.001. Epub 2015 Dec 7. Prog Nucl Magn Reson Spectrosc. 2016. PMID: 26952190 Free PMC article. Review.
-
Enzyme dynamics from NMR spectroscopy.Acc Chem Res. 2015 Feb 17;48(2):457-65. doi: 10.1021/ar500340a. Epub 2015 Jan 9. Acc Chem Res. 2015. PMID: 25574774 Free PMC article. Review.
-
Solution NMR and computational methods for understanding protein allostery.J Phys Chem B. 2013 Mar 21;117(11):3063-73. doi: 10.1021/jp312576v. Epub 2013 Mar 12. J Phys Chem B. 2013. PMID: 23445323 Free PMC article.
Cited by
-
Intrinsic local destabilization of the C-terminus predisposes integrin α1 I domain to a conformational switch induced by collagen binding.Protein Sci. 2016 Sep;25(9):1672-81. doi: 10.1002/pro.2972. Epub 2016 Aug 1. Protein Sci. 2016. PMID: 27342747 Free PMC article.
-
Ensemble origins and distance-dependence of long-range mutational effects in proteins.iScience. 2022 Sep 22;25(10):105181. doi: 10.1016/j.isci.2022.105181. eCollection 2022 Oct 21. iScience. 2022. PMID: 36248733 Free PMC article.
-
Thermodynamic and NMR Assessment of Ligand Cooperativity and Intersubunit Communication in Symmetric Dimers: Application to Thymidylate Synthase.Front Mol Biosci. 2018 May 25;5:47. doi: 10.3389/fmolb.2018.00047. eCollection 2018. Front Mol Biosci. 2018. PMID: 29888227 Free PMC article. Review.
-
High-fidelity, hyper-accurate, and evolved mutants rewire atomic-level communication in CRISPR-Cas9.Sci Adv. 2024 Mar 8;10(10):eadl1045. doi: 10.1126/sciadv.adl1045. Epub 2024 Mar 6. Sci Adv. 2024. PMID: 38446895 Free PMC article.
-
Allosteric inhibition explained through conformational ensembles sampling distinct "mixed" states.Comput Struct Biotechnol J. 2020 Nov 11;18:3803-3818. doi: 10.1016/j.csbj.2020.10.026. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 33335680 Free PMC article. Review.
References
-
- Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965;12:88–118. - PubMed
-
- Koshland DE, Jr., Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
