

Disturbances of Ligand Potency and Enhanced Degradation of the Human Glycine Receptor at Affected Positions G160 and T162 Originally Identified in Patients Suffering from Hyperekplexia
- PMID: 26733802
- PMCID: PMC4686643
- DOI: 10.3389/fnmol.2015.00079
Disturbances of Ligand Potency and Enhanced Degradation of the Human Glycine Receptor at Affected Positions G160 and T162 Originally Identified in Patients Suffering from Hyperekplexia
Abstract
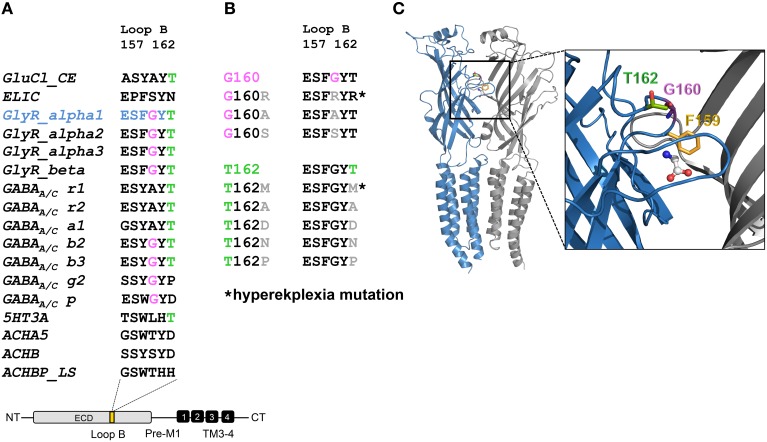
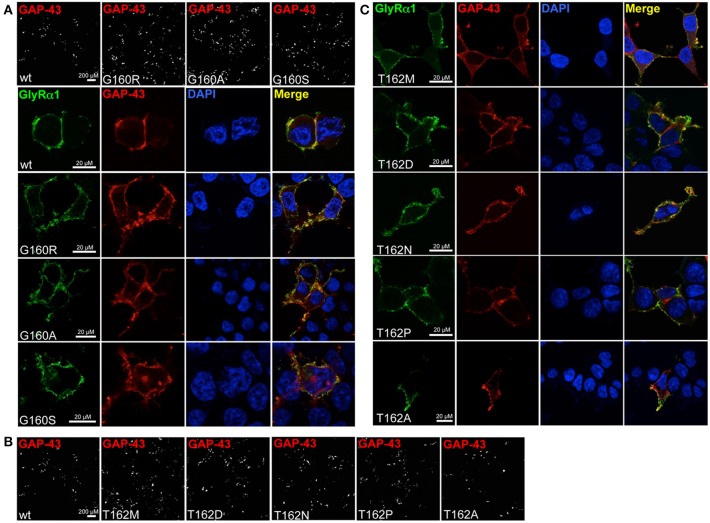
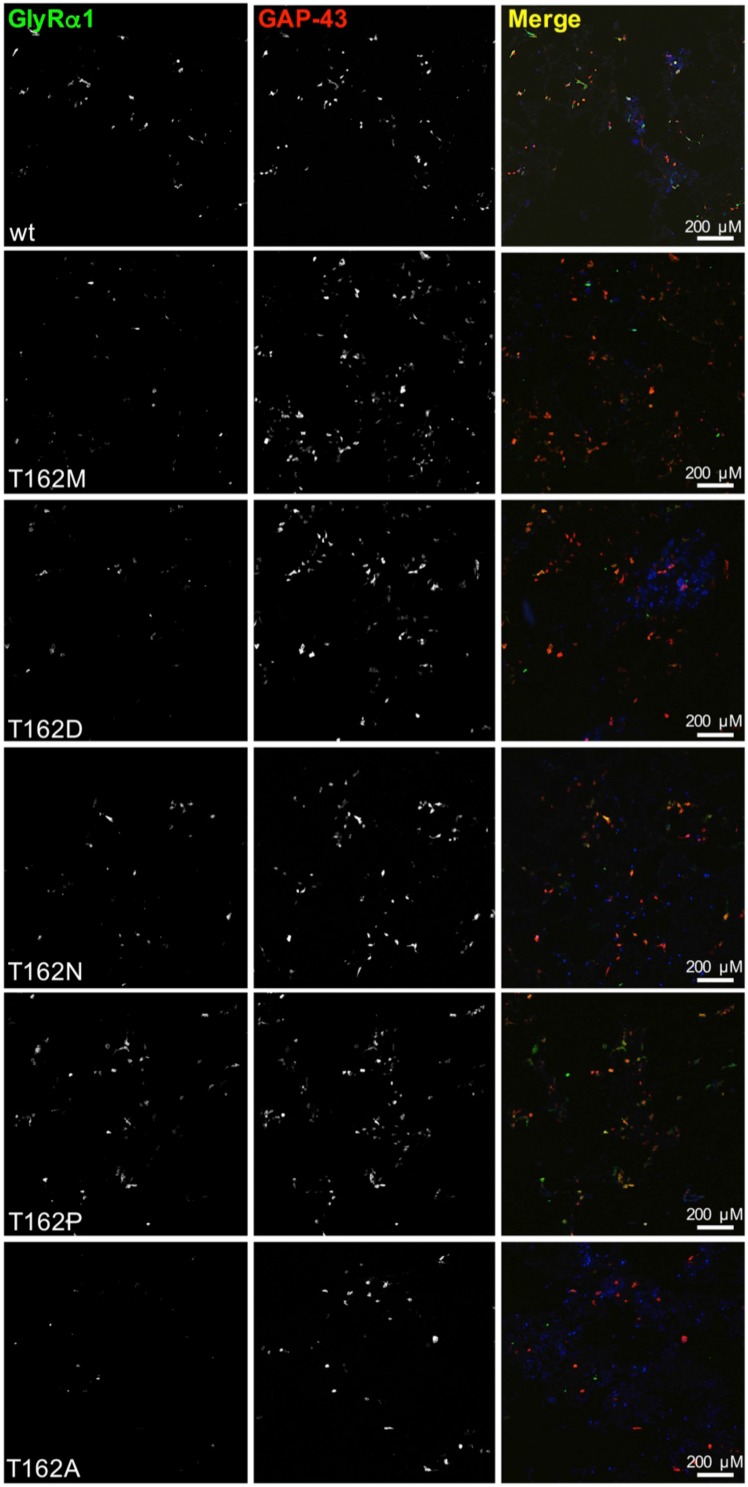
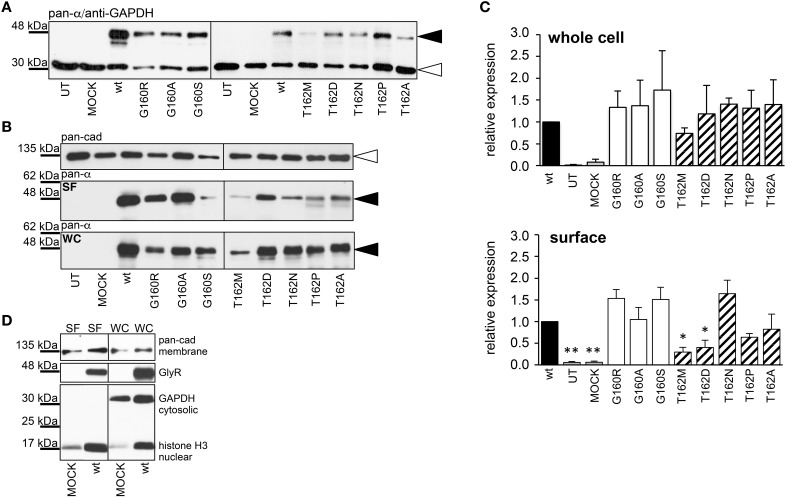
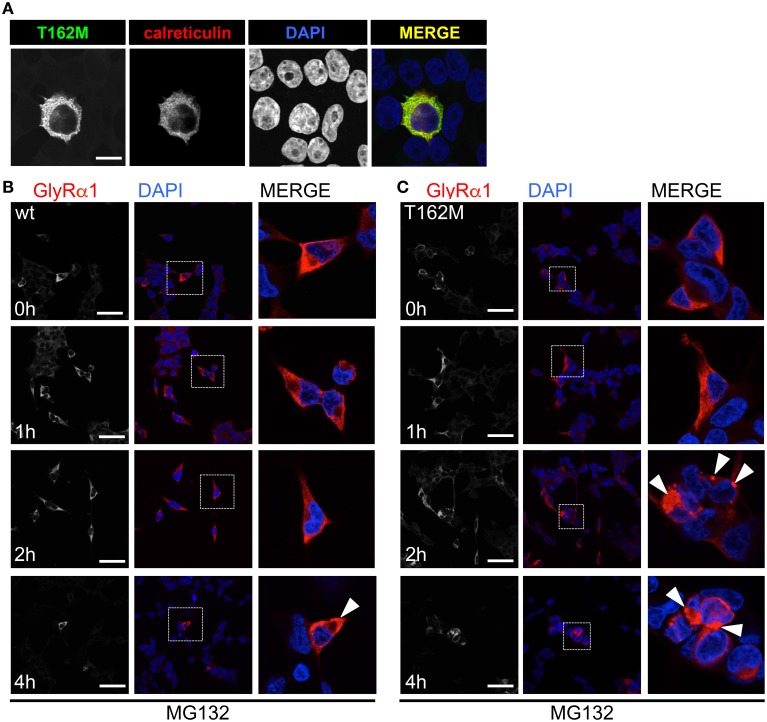
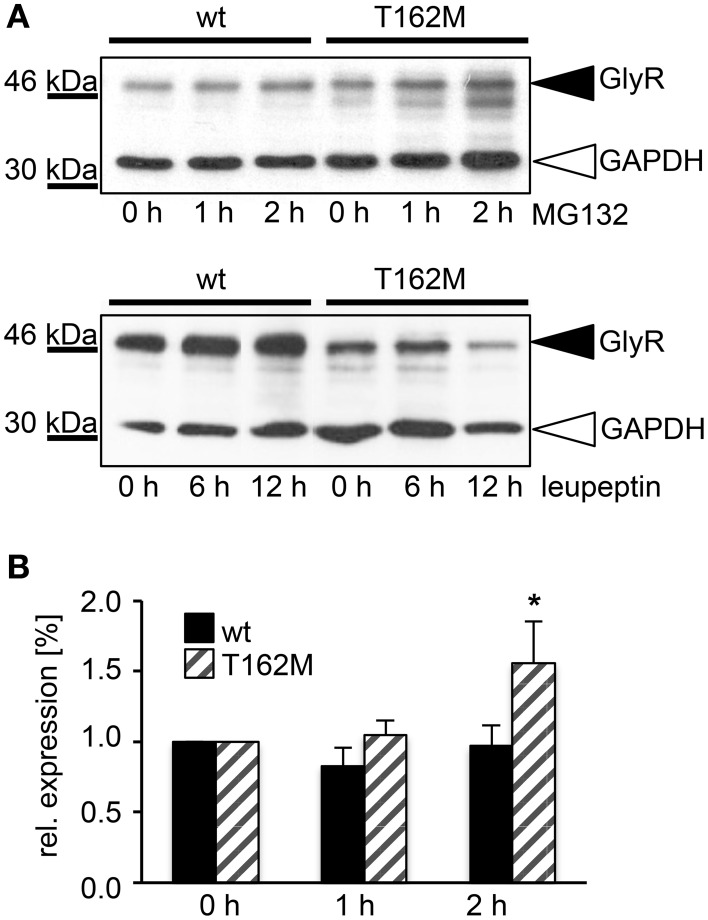
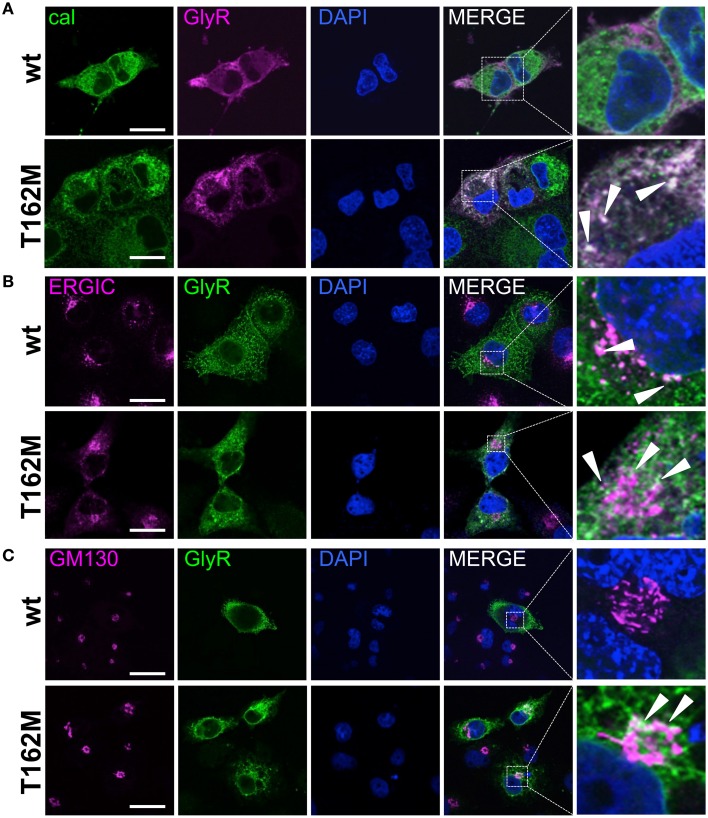
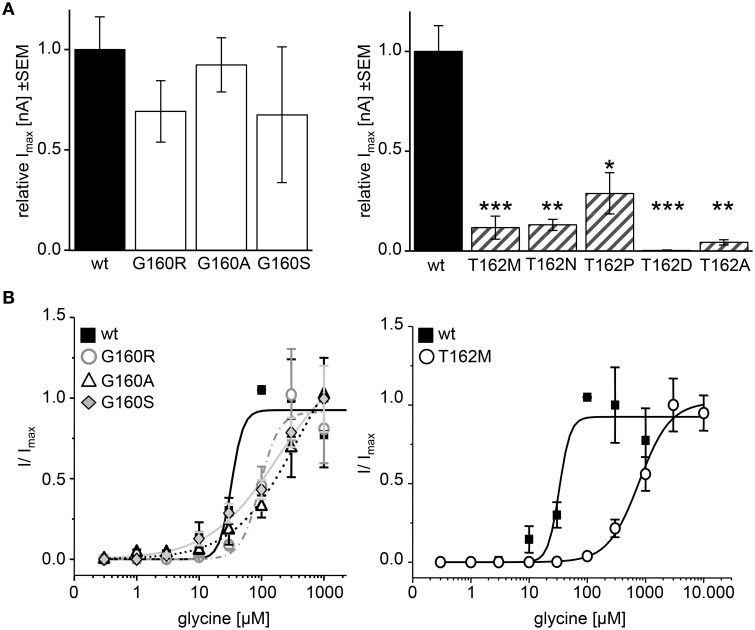
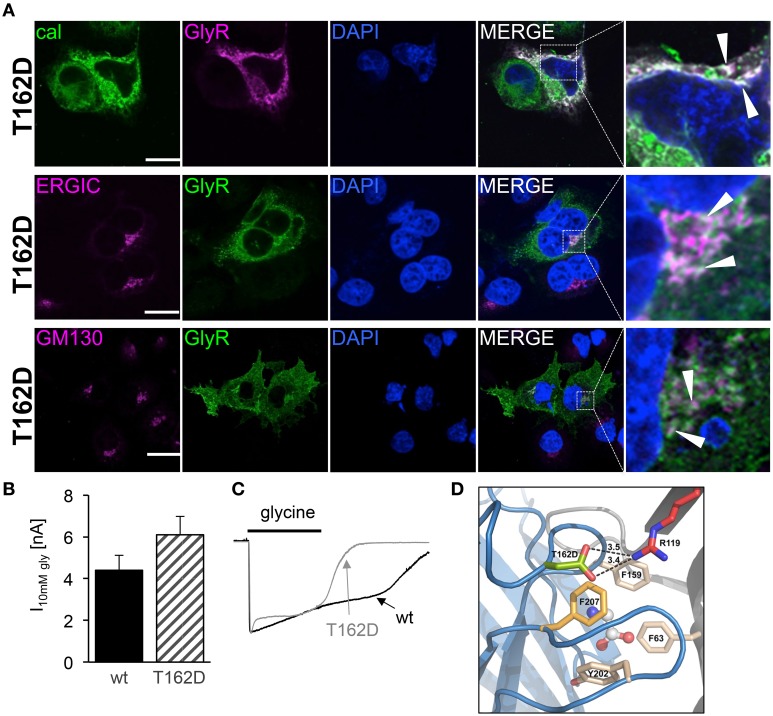
Ligand-binding of Cys-loop receptors is determined by N-terminal extracellular loop structures from the plus as well as from the minus side of two adjacent subunits in the pentameric receptor complex. An aromatic residue in loop B of the glycine receptor (GlyR) undergoes direct interaction with the incoming ligand via a cation-π interaction. Recently, we showed that mutated residues in loop B identified from human patients suffering from hyperekplexia disturb ligand-binding. Here, we exchanged the affected human residues by amino acids found in related members of the Cys-loop receptor family to determine the effects of side chain volume for ion channel properties. GlyR variants were characterized in vitro following transfection into cell lines in order to analyze protein expression, trafficking, degradation and ion channel function. GlyR α1 G160 mutations significantly decrease glycine potency arguing for a positional effect on neighboring aromatic residues and consequently glycine-binding within the ligand-binding pocket. Disturbed glycinergic inhibition due to T162 α1 mutations is an additive effect of affected biogenesis and structural changes within the ligand-binding site. Protein trafficking from the ER toward the ER-Golgi intermediate compartment, the secretory Golgi pathways and finally the cell surface is largely diminished, but still sufficient to deliver ion channels that are functional at least at high glycine concentrations. The majority of T162 mutant protein accumulates in the ER and is delivered to ER-associated proteasomal degradation. Hence, G160 is an important determinant during glycine binding. In contrast, T162 affects primarily receptor biogenesis whereas exchanges in functionality are secondary effects thereof.
Keywords: Cys-loop receptor; glycine receptor; hyperekplexia; ligand potencies; loop B; side chain properties.
Figures
Similar articles
-
The GlyR Extracellular β8-β9 Loop - A Functional Determinant of Agonist Potency.Front Mol Neurosci. 2017 Oct 9;10:322. doi: 10.3389/fnmol.2017.00322. eCollection 2017. Front Mol Neurosci. 2017. PMID: 29062270 Free PMC article.
-
Impaired Glycine Receptor Trafficking in Neurological Diseases.Front Mol Neurosci. 2018 Aug 21;11:291. doi: 10.3389/fnmol.2018.00291. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30186111 Free PMC article. Review.
-
Disturbed neuronal ER-Golgi sorting of unassembled glycine receptors suggests altered subcellular processing is a cause of human hyperekplexia.J Neurosci. 2015 Jan 7;35(1):422-37. doi: 10.1523/JNEUROSCI.1509-14.2015. J Neurosci. 2015. PMID: 25568133 Free PMC article.
-
Charged residues at the pore extracellular half of the glycine receptor facilitate channel gating: a potential role played by electrostatic repulsion.J Physiol. 2020 Oct;598(20):4643-4661. doi: 10.1113/JP279288. Epub 2020 Aug 26. J Physiol. 2020. PMID: 32844405
-
Structural insights into Cys-loop receptor function and ligand recognition.Biochem Pharmacol. 2013 Oct 15;86(8):1042-53. doi: 10.1016/j.bcp.2013.07.001. Epub 2013 Jul 10. Biochem Pharmacol. 2013. PMID: 23850718 Review.
Cited by
-
The GlyR Extracellular β8-β9 Loop - A Functional Determinant of Agonist Potency.Front Mol Neurosci. 2017 Oct 9;10:322. doi: 10.3389/fnmol.2017.00322. eCollection 2017. Front Mol Neurosci. 2017. PMID: 29062270 Free PMC article.
-
Impaired Glycine Receptor Trafficking in Neurological Diseases.Front Mol Neurosci. 2018 Aug 21;11:291. doi: 10.3389/fnmol.2018.00291. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30186111 Free PMC article. Review.
-
Glycine receptor autoantibody binding to the extracellular domain is independent from receptor glycosylation.Front Mol Neurosci. 2023 Feb 13;16:1089101. doi: 10.3389/fnmol.2023.1089101. eCollection 2023. Front Mol Neurosci. 2023. PMID: 36860666 Free PMC article.
-
Disruption of a Structurally Important Extracellular Element in the Glycine Receptor Leads to Decreased Synaptic Integration and Signaling Resulting in Severe Startle Disease.J Neurosci. 2017 Aug 16;37(33):7948-7961. doi: 10.1523/JNEUROSCI.0009-17.2017. Epub 2017 Jul 19. J Neurosci. 2017. PMID: 28724750 Free PMC article.
-
Novel Functional Properties of Missense Mutations in the Glycine Receptor β Subunit in Startle Disease.Front Mol Neurosci. 2021 Sep 24;14:745275. doi: 10.3389/fnmol.2021.745275. eCollection 2021. Front Mol Neurosci. 2021. PMID: 34630038 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous