

Tau Oligomers: The Toxic Player at Synapses in Alzheimer's Disease
- PMID: 26696824
- PMCID: PMC4667007
- DOI: 10.3389/fncel.2015.00464
Tau Oligomers: The Toxic Player at Synapses in Alzheimer's Disease
Abstract
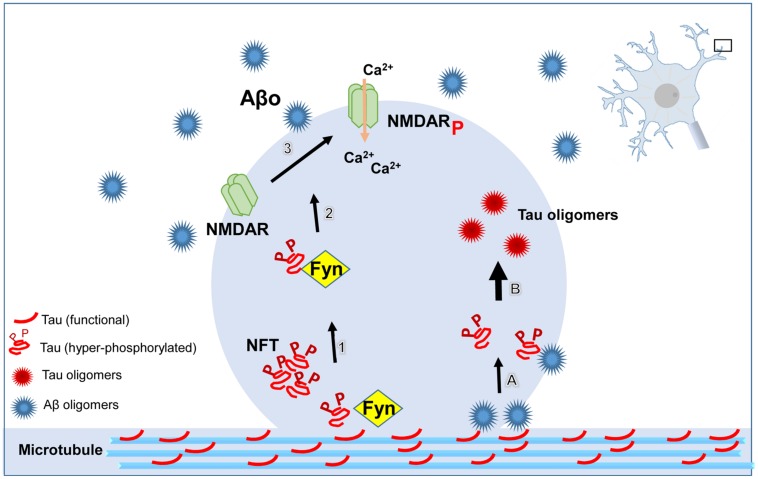
Alzheimer's disease (AD) is a progressive disorder in which the most noticeable symptoms are cognitive impairment and memory loss. However, the precise mechanism by which those symptoms develop remains unknown. Of note, neuronal loss occurs at sites where synaptic dysfunction is observed earlier, suggesting that altered synaptic connections precede neuronal loss. The abnormal accumulation of amyloid-β (Aβ) and tau protein is the main histopathological feature of the disease. Several lines of evidence suggest that the small oligomeric forms of Aβ and tau may act synergistically to promote synaptic dysfunction in AD. Remarkably, tau pathology correlates better with the progression of the disease than Aβ. Recently, a growing number of studies have begun to suggest that missorting of tau protein from the axon to the dendrites is required to mediate the detrimental effects of Aβ. In this review we discuss the novel findings regarding the potential mechanisms by which tau oligomers contribute to synaptic dysfunction in AD.
Keywords: Alzheimer’s disease; Aβ oligomers; dendrites; synapsis; tau oligomers.
Figures
Similar articles
-
Synaptic Dysfunction in Alzheimer's Disease: Aβ, Tau, and Epigenetic Alterations.Mol Neurobiol. 2018 Apr;55(4):3021-3032. doi: 10.1007/s12035-017-0533-3. Epub 2017 Apr 29. Mol Neurobiol. 2018. PMID: 28456942 Review.
-
Linking amyloid-β and tau: amyloid-β induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton.Neurodegener Dis. 2012;10(1-4):64-72. doi: 10.1159/000332816. Epub 2011 Dec 7. Neurodegener Dis. 2012. PMID: 22156588
-
Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer's disease model mice.Life Sci. 2012 Dec 10;91(23-24):1169-76. doi: 10.1016/j.lfs.2011.12.022. Epub 2012 Jan 17. Life Sci. 2012. PMID: 22273754
-
Dendritic/Post-synaptic Tau and Early Pathology of Alzheimer's Disease.Front Mol Neurosci. 2021 Jun 25;14:671779. doi: 10.3389/fnmol.2021.671779. eCollection 2021. Front Mol Neurosci. 2021. PMID: 34248498 Free PMC article. Review.
-
Synaptic Amyloid-β Oligomers Precede p-Tau and Differentiate High Pathology Control Cases.Am J Pathol. 2016 Jan;186(1):185-98. doi: 10.1016/j.ajpath.2015.09.018. Am J Pathol. 2016. PMID: 26718979 Free PMC article.
Cited by
-
Advances in protein misfolding, amyloidosis and its correlation with human diseases.3 Biotech. 2020 May;10(5):193. doi: 10.1007/s13205-020-2166-x. Epub 2020 Apr 4. 3 Biotech. 2020. PMID: 32269898 Free PMC article. Review.
-
Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons.Glia. 2017 Aug;65(8):1302-1316. doi: 10.1002/glia.23163. Epub 2017 May 18. Glia. 2017. PMID: 28519902 Free PMC article.
-
Tau-Reactive Endogenous Antibodies: Origin, Functionality, and Implications for the Pathophysiology of Alzheimer's Disease.J Immunol Res. 2019 Aug 6;2019:7406810. doi: 10.1155/2019/7406810. eCollection 2019. J Immunol Res. 2019. PMID: 31687413 Free PMC article. Review.
-
Intrathecal amyloid-beta oligomer administration increases tau phosphorylation in the medial temporal lobe in the African green monkey: A nonhuman primate model of Alzheimer's disease.Neuropathol Appl Neurobiol. 2022 Jun;48(4):e12800. doi: 10.1111/nan.12800. Epub 2022 Mar 2. Neuropathol Appl Neurobiol. 2022. PMID: 35156715 Free PMC article.
-
The role of pathological tau in synaptic dysfunction in Alzheimer's diseases.Transl Neurodegener. 2021 Nov 10;10(1):45. doi: 10.1186/s40035-021-00270-1. Transl Neurodegener. 2021. PMID: 34753506 Free PMC article. Review.
References
-
- Ahmed Z., Cooper J., Murray T., Garn K., McNaughton E., Clarke H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127 667–683. 10.1007/s00401-014-1254-6 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources