

Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1
- PMID: 26676772
- PMCID: PMC4810695
- DOI: 10.1128/JVI.02749-15
Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1
Abstract
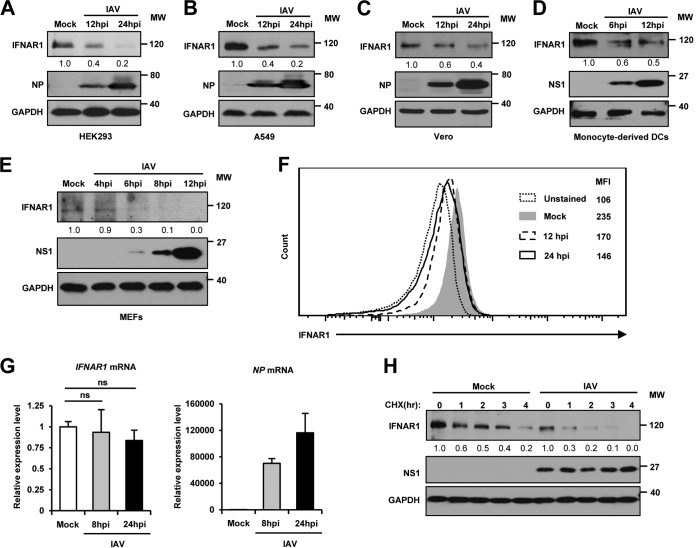
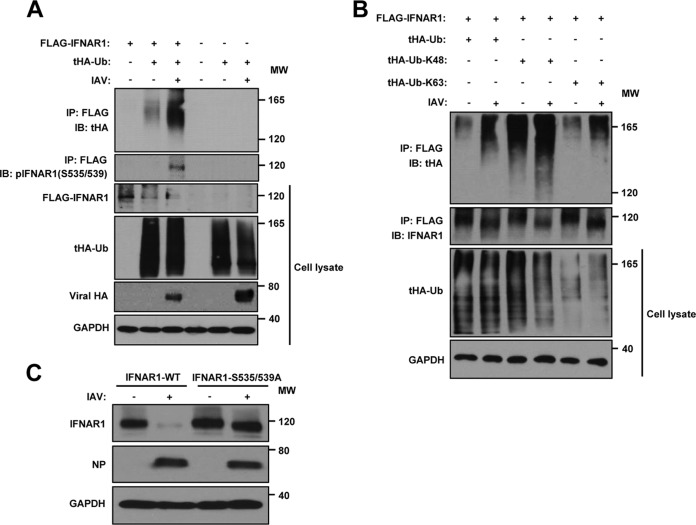
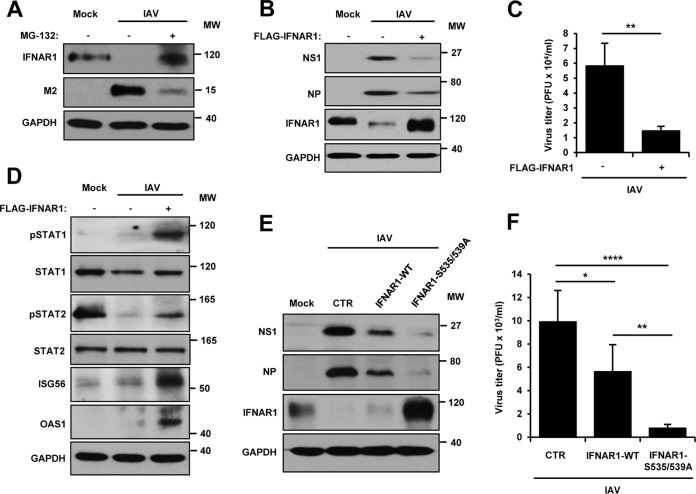
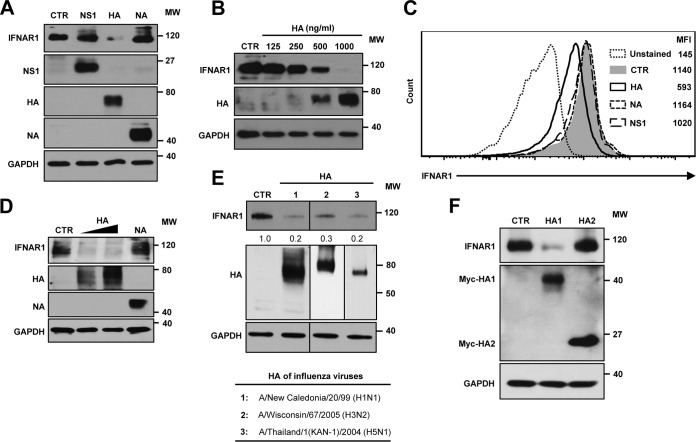
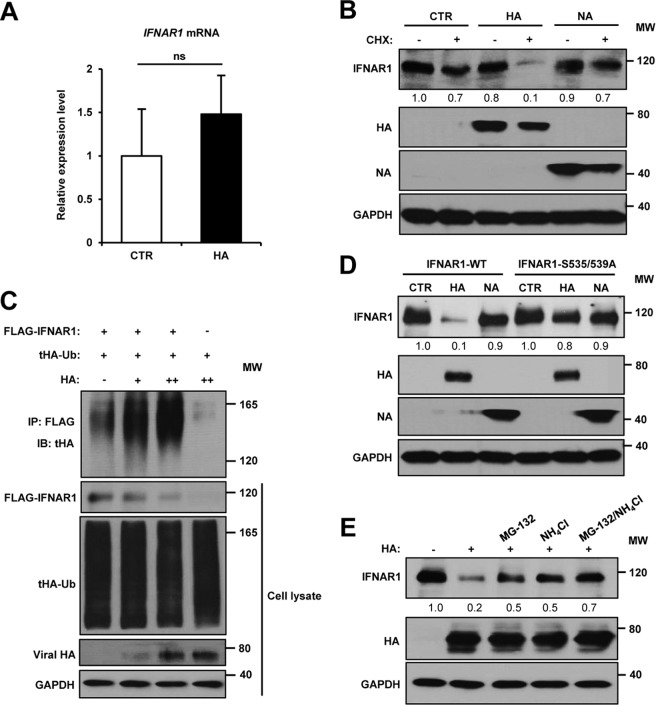
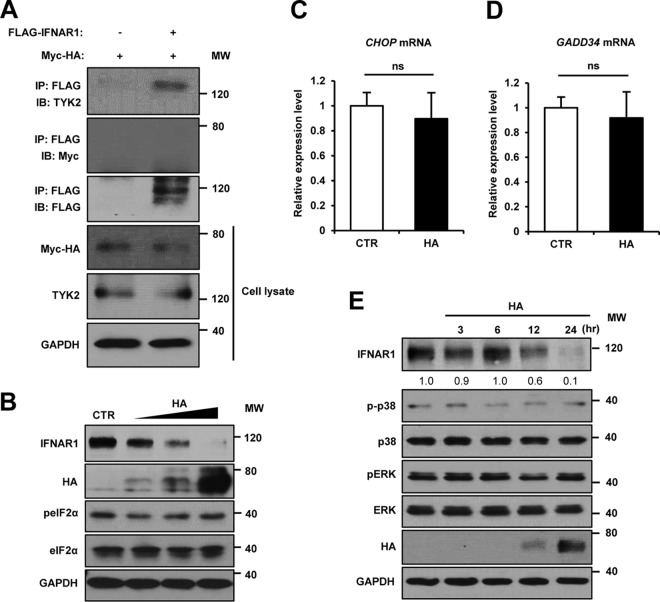
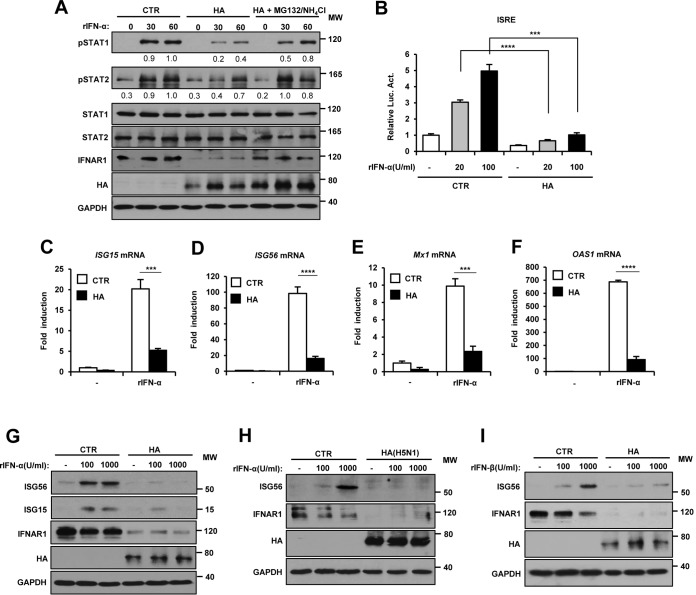
Influenza A virus (IAV) employs diverse strategies to circumvent type I interferon (IFN) responses, particularly by inhibiting the synthesis of type I IFNs. However, it is poorly understood if and how IAV regulates the type I IFN receptor (IFNAR)-mediated signaling mode. In this study, we demonstrate that IAV induces the degradation of IFNAR subunit 1 (IFNAR1) to attenuate the type I IFN-induced antiviral signaling pathway. Following infection, the level of IFNAR1 protein, but not mRNA, decreased. Indeed, IFNAR1 was phosphorylated and ubiquitinated by IAV infection, which resulted in IFNAR1 elimination. The transiently overexpressed IFNAR1 displayed antiviral activity by inhibiting virus replication. Importantly, the hemagglutinin (HA) protein of IAV was proved to trigger the ubiquitination of IFNAR1, diminishing the levels of IFNAR1. Further, influenza A viral HA1 subunit, but not HA2 subunit, downregulated IFNAR1. However, viral HA-mediated degradation of IFNAR1 was not caused by the endoplasmic reticulum (ER) stress response. IAV HA robustly reduced cellular sensitivity to type I IFNs, suppressing the activation of STAT1/STAT2 and induction of IFN-stimulated antiviral proteins. Taken together, our findings suggest that IAV HA causes IFNAR1 degradation, which in turn helps the virus escape the powerful innate immune system. Thus, the research elucidated an influenza viral mechanism for eluding the IFNAR signaling pathway, which could provide new insights into the interplay between influenza virus and host innate immunity.
Importance: Influenza A virus (IAV) infection causes significant morbidity and mortality worldwide and remains a major health concern. When triggered by influenza viral infection, host cells produce type I interferon (IFN) to block viral replication. Although IAV was shown to have diverse strategies to evade this powerful, IFN-mediated antiviral response, it is not well-defined if IAV manipulates the IFN receptor-mediated signaling pathway. Here, we uncovered that influenza viral hemagglutinin (HA) protein causes the degradation of type I IFN receptor subunit 1 (IFNAR1). HA promoted phosphorylation and polyubiquitination of IFNAR1, which facilitated the degradation of this receptor. The HA-mediated elimination of IFNAR1 notably decreased the cells' sensitivities to type I IFNs, as demonstrated by the diminished expression of IFN-induced antiviral genes. This discovery could help us understand how IAV regulates the host innate immune response to create an environment optimized for viral survival in host cells.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Casein Kinase 1α Mediates the Degradation of Receptors for Type I and Type II Interferons Caused by Hemagglutinin of Influenza A Virus.J Virol. 2018 Mar 14;92(7):e00006-18. doi: 10.1128/JVI.00006-18. Print 2018 Apr 1. J Virol. 2018. PMID: 29343571 Free PMC article.
-
PARP1 Enhances Influenza A Virus Propagation by Facilitating Degradation of Host Type I Interferon Receptor.J Virol. 2020 Mar 17;94(7):e01572-19. doi: 10.1128/JVI.01572-19. Print 2020 Mar 17. J Virol. 2020. PMID: 31915279 Free PMC article.
-
Innate Sensing of Influenza A Virus Hemagglutinin Glycoproteins by the Host Endoplasmic Reticulum (ER) Stress Pathway Triggers a Potent Antiviral Response via ER-Associated Protein Degradation.J Virol. 2017 Dec 14;92(1):e01690-17. doi: 10.1128/JVI.01690-17. Print 2018 Jan 1. J Virol. 2017. PMID: 29046440 Free PMC article.
-
Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins.Viruses. 2018 Dec 12;10(12):708. doi: 10.3390/v10120708. Viruses. 2018. PMID: 30545063 Free PMC article. Review.
-
The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains?J Innate Immun. 2020;12(6):437-447. doi: 10.1159/000508379. Epub 2020 Jun 19. J Innate Immun. 2020. PMID: 32564033 Free PMC article. Review.
Cited by
-
Role of Post-translational Modifications in Influenza A Virus Life Cycle and Host Innate Immune Response.Front Microbiol. 2020 Sep 4;11:517461. doi: 10.3389/fmicb.2020.517461. eCollection 2020. Front Microbiol. 2020. PMID: 33013775 Free PMC article. Review.
-
Differential Responsiveness of Monocyte and Macrophage Subsets to Interferon.Arthritis Rheumatol. 2020 Jan;72(1):100-113. doi: 10.1002/art.41072. Epub 2019 Dec 10. Arthritis Rheumatol. 2020. PMID: 31390156 Free PMC article.
-
Induction and Subversion of Human Protective Immunity: Contrasting Influenza and Respiratory Syncytial Virus.Front Immunol. 2018 Mar 2;9:323. doi: 10.3389/fimmu.2018.00323. eCollection 2018. Front Immunol. 2018. PMID: 29552008 Free PMC article. Review.
-
Herpes Simplex Virus 1 UL36USP Antagonizes Type I Interferon-Mediated Antiviral Innate Immunity.J Virol. 2018 Sep 12;92(19):e01161-18. doi: 10.1128/JVI.01161-18. Print 2018 Oct 1. J Virol. 2018. PMID: 29997210 Free PMC article.
-
Pseudorabies Virus dUTPase UL50 Induces Lysosomal Degradation of Type I Interferon Receptor 1 and Antagonizes the Alpha Interferon Response.J Virol. 2017 Oct 13;91(21):e01148-17. doi: 10.1128/JVI.01148-17. Print 2017 Nov 1. J Virol. 2017. PMID: 28794045 Free PMC article.
References
-
- D'Mello T, Brammer L, Blanton L, Kniss K, Smith S, Mustaquim D, Steffens C, Dhara R, Cohen J, Chaves SS, Finelli L, Bresee J, Wallis T, Xu X, Abd Elal AI, Gubareva L, Wentworth D, Villanueva J, Katz J, Jernigan D, Centers for Disease Control and Prevention. 2015. Update: influenza activity—United States, September 28, 2014-February 21, 2015. MMWR Morb Mortal Wkly Rep 64:206–212. - PMC - PubMed
-
- Cowling BJ, Jin L, Lau EH, Liao Q, Wu P, Jiang H, Tsang TK, Zheng J, Fang VJ, Chang Z, Ni MY, Zhang Q, Ip DK, Yu J, Li Y, Wang L, Tu W, Meng L, Wu JT, Luo H, Li Q, Shu Y, Li Z, Feng Z, Yang W, Wang Y, Leung GM, Yu H. 2013. Comparative epidemiology of human infections with avian influenza A H7N9 and H5N1 viruses in China: a population-based study of laboratory-confirmed cases. Lancet 382:129–137. doi:10.1016/S0140-6736(13)61171-X. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
