

cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins
- PMID: 26669444
- PMCID: PMC4702992
- DOI: 10.1073/pnas.1522332112
cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins
Abstract
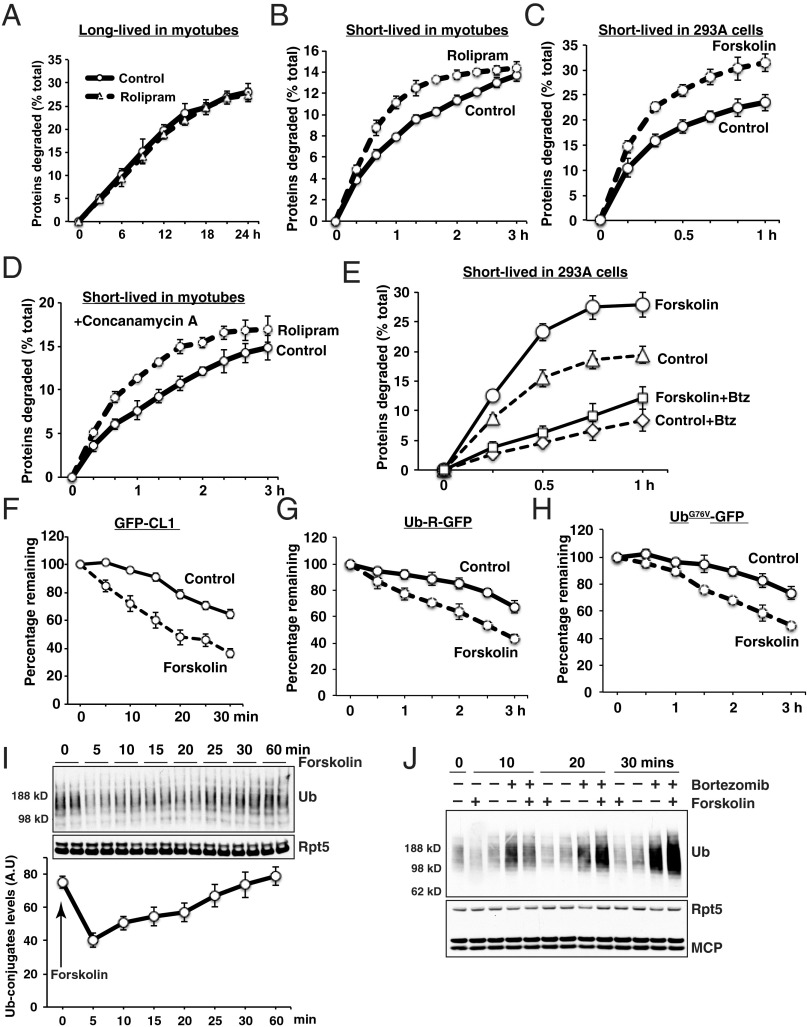
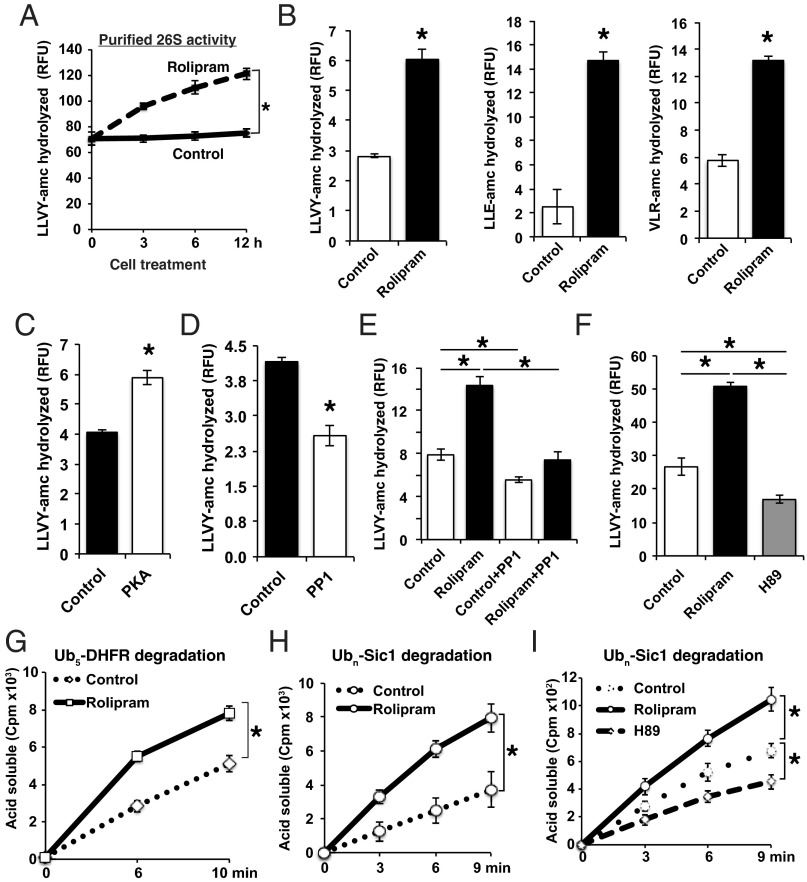
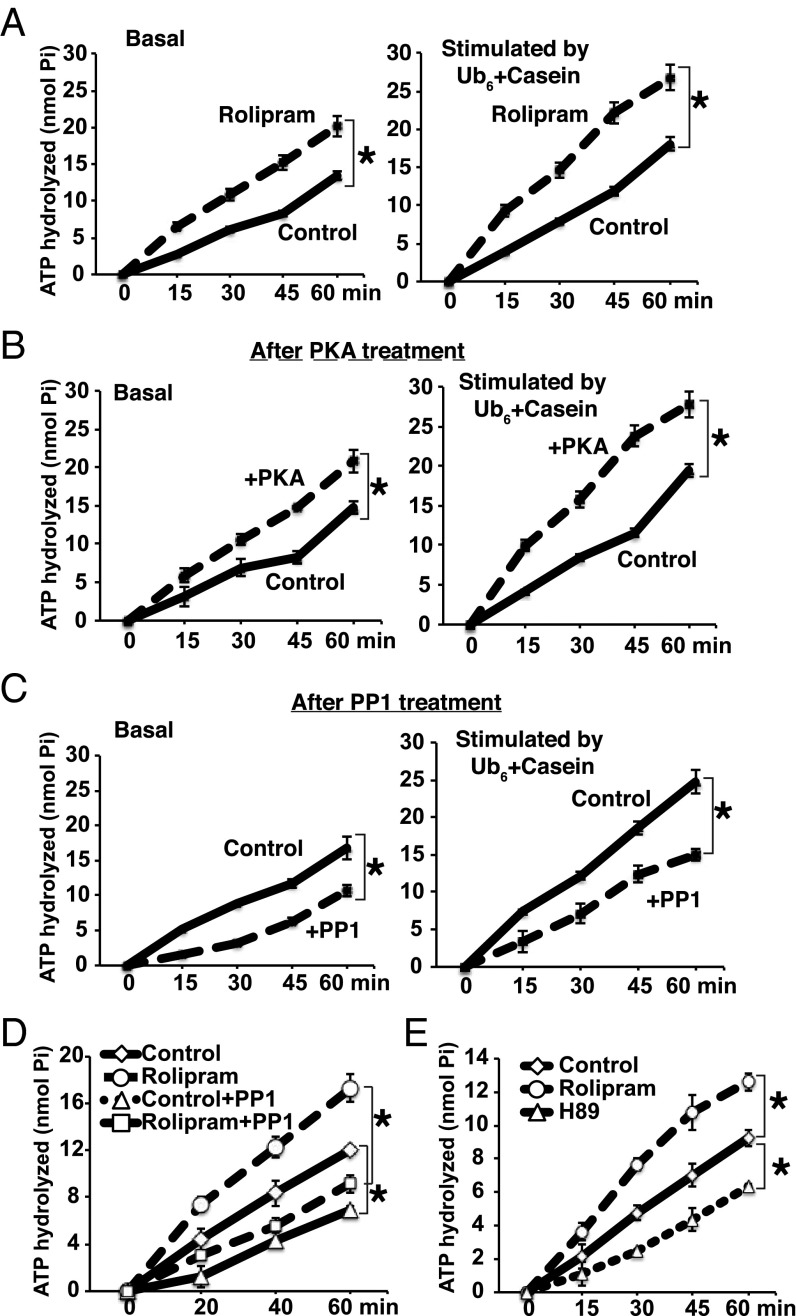
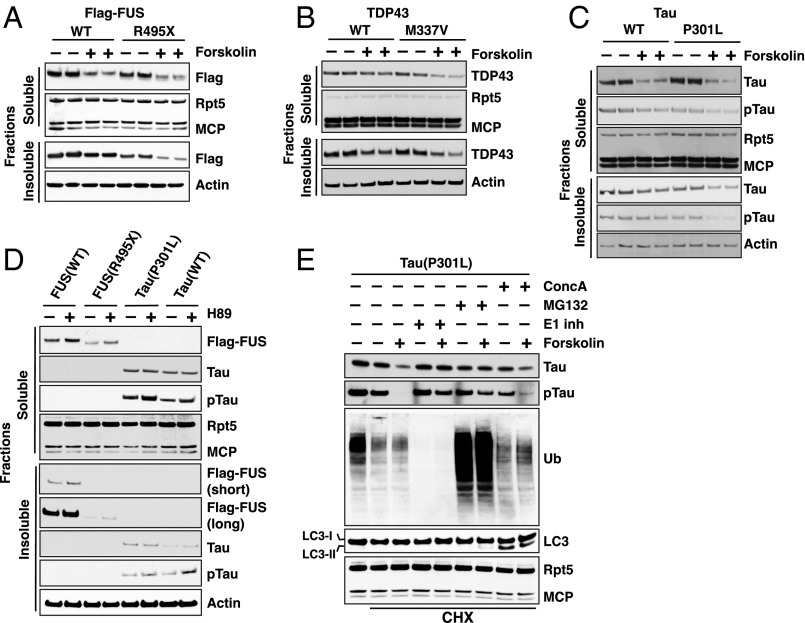
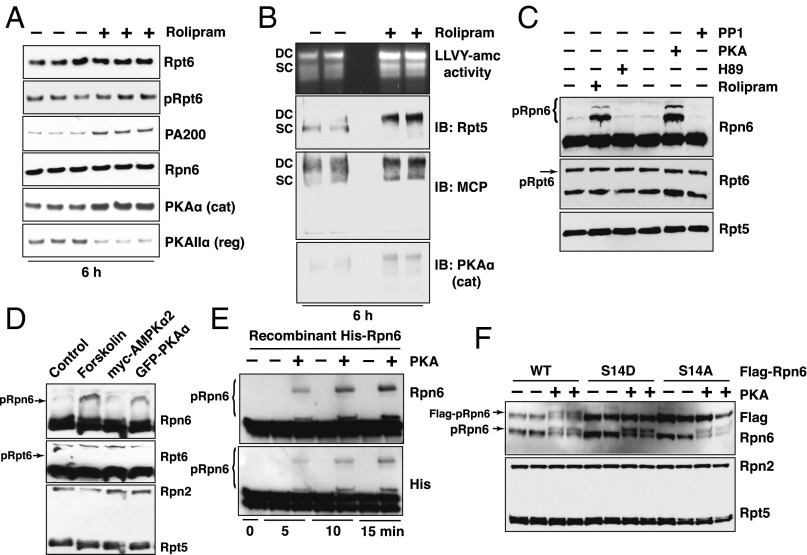
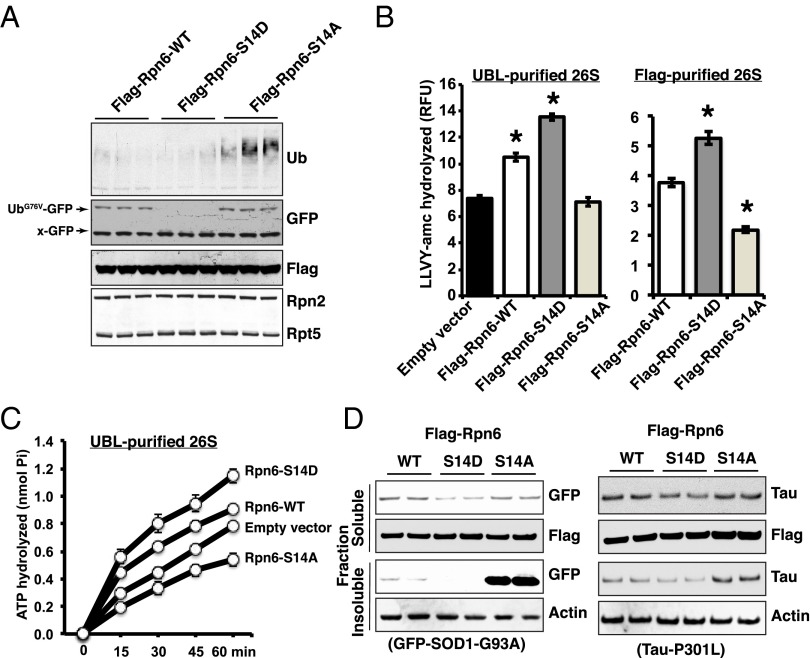
Although rates of protein degradation by the ubiquitin-proteasome pathway (UPS) are determined by their rates of ubiquitination, we show here that the proteasome's capacity to degrade ubiquitinated proteins is also tightly regulated. We studied the effects of cAMP-dependent protein kinase (PKA) on proteolysis by the UPS in several mammalian cell lines. Various agents that raise intracellular cAMP and activate PKA (activators of adenylate cyclase or inhibitors of phosphodiesterase 4) promoted degradation of short-lived (but not long-lived) cell proteins generally, model UPS substrates having different degrons, and aggregation-prone proteins associated with major neurodegenerative diseases, including mutant FUS (Fused in sarcoma), SOD1 (superoxide dismutase 1), TDP43 (TAR DNA-binding protein 43), and tau. 26S proteasomes purified from these treated cells or from control cells and treated with PKA degraded ubiquitinated proteins, small peptides, and ATP more rapidly than controls, but not when treated with protein phosphatase. Raising cAMP levels also increased amounts of doubly capped 26S proteasomes. Activated PKA phosphorylates the 19S subunit, Rpn6/PSMD11 (regulatory particle non-ATPase 6/proteasome subunit D11) at Ser14. Overexpression of a phosphomimetic Rpn6 mutant activated proteasomes similarly, whereas a nonphosphorylatable mutant decreased activity. Thus, proteasome function and protein degradation are regulated by cAMP through PKA and Rpn6, and activation of proteasomes by this mechanism may be useful in treating proteotoxic diseases.
Keywords: Rpn6/PSMD11; cAMP; cAMP-dependent protein kinase; proteasomes; protein degradation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
26S Proteasomes are rapidly activated by diverse hormones and physiological states that raise cAMP and cause Rpn6 phosphorylation.Proc Natl Acad Sci U S A. 2019 Mar 5;116(10):4228-4237. doi: 10.1073/pnas.1809254116. Epub 2019 Feb 19. Proc Natl Acad Sci U S A. 2019. PMID: 30782827 Free PMC article.
-
Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling.Nat Med. 2016 Jan;22(1):46-53. doi: 10.1038/nm.4011. Epub 2015 Dec 21. Nat Med. 2016. PMID: 26692334 Free PMC article.
-
cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases.Proc Natl Acad Sci U S A. 2020 Jun 23;117(25):14220-14230. doi: 10.1073/pnas.2003277117. Epub 2020 Jun 8. Proc Natl Acad Sci U S A. 2020. PMID: 32513741 Free PMC article.
-
Mechanisms That Activate 26S Proteasomes and Enhance Protein Degradation.Biomolecules. 2021 May 22;11(6):779. doi: 10.3390/biom11060779. Biomolecules. 2021. PMID: 34067263 Free PMC article. Review.
-
Regulating protein breakdown through proteasome phosphorylation.Biochem J. 2017 Sep 24;474(19):3355-3371. doi: 10.1042/BCJ20160809. Biochem J. 2017. PMID: 28947610 Free PMC article. Review.
Cited by
-
Protein Degradation and the Pathologic Basis of Disease.Am J Pathol. 2019 Jan;189(1):94-103. doi: 10.1016/j.ajpath.2018.09.004. Epub 2018 Oct 10. Am J Pathol. 2019. PMID: 30312581 Free PMC article. Review.
-
Molecular mechanism for activation of the 26S proteasome by ZFAND5.Mol Cell. 2023 Aug 17;83(16):2959-2975.e7. doi: 10.1016/j.molcel.2023.07.023. Mol Cell. 2023. PMID: 37595557 Free PMC article.
-
Novel Therapies for Parkinsonian Syndromes-Recent Progress and Future Perspectives.Front Mol Neurosci. 2021 Aug 26;14:720220. doi: 10.3389/fnmol.2021.720220. eCollection 2021. Front Mol Neurosci. 2021. PMID: 34512258 Free PMC article. Review.
-
Early deficits in an in vitro striatal microcircuit model carrying the Parkinson's GBA-N370S mutation.NPJ Parkinsons Dis. 2024 Apr 12;10(1):82. doi: 10.1038/s41531-024-00694-2. NPJ Parkinsons Dis. 2024. PMID: 38609392 Free PMC article.
-
Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers.Proc Natl Acad Sci U S A. 2017 Jan 10;114(2):382-387. doi: 10.1073/pnas.1619067114. Epub 2016 Dec 27. Proc Natl Acad Sci U S A. 2017. PMID: 28028240 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
