

Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function
- PMID: 26660451
- PMCID: PMC5446573
- DOI: 10.1111/bph.13403
Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function
Abstract
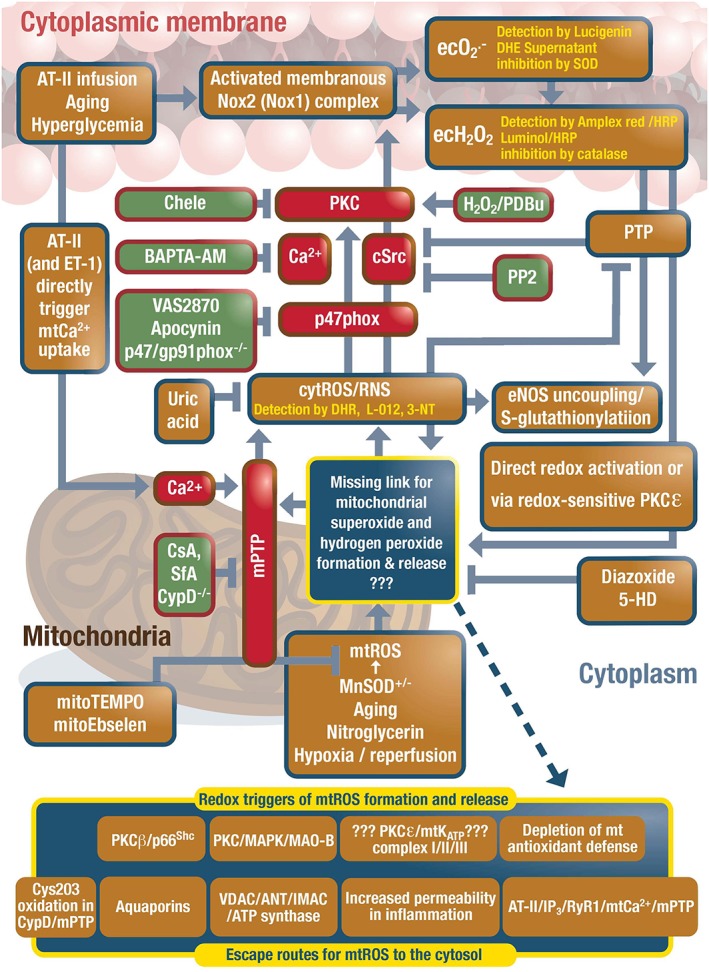
Cardiovascular diseases are associated with and/or caused by oxidative stress. This concept has been proven by using the approach of genetic deletion of reactive species producing (pro-oxidant) enzymes as well as by the overexpression of reactive species detoxifying (antioxidant) enzymes leading to a marked reduction of reactive oxygen and nitrogen species (RONS) and in parallel to an amelioration of the severity of diseases. Likewise, the development and progression of cardiovascular diseases is aggravated by overexpression of RONS producing enzymes as well as deletion of antioxidant RONS detoxifying enzymes. Thus, the consequences of the interaction (redox crosstalk) of superoxide/hydrogen peroxide produced by mitochondria with other ROS producing enzymes such as NADPH oxidases (Nox) are of outstanding importance and will be discussed including the consequences for endothelial nitric oxide synthase (eNOS) uncoupling as well as the redox regulation of the vascular function/tone in general (soluble guanylyl cyclase, endothelin-1, prostanoid synthesis). Pathways and potential mechanisms leading to this crosstalk will be analysed in detail and highlighted by selected examples from the current literature including hypoxia, angiotensin II-induced hypertension, nitrate tolerance, aging and others. The general concept of redox-based activation of RONS sources via "kindling radicals" and enzyme-specific "redox switches" will be discussed providing evidence that mitochondria represent key players and amplifiers of the burden of oxidative stress.
Linked articles: This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc.
© 2015 The British Pharmacological Society.
Figures
Similar articles
-
Regulation of Vascular Function and Inflammation via Cross Talk of Reactive Oxygen and Nitrogen Species from Mitochondria or NADPH Oxidase-Implications for Diabetes Progression.Int J Mol Sci. 2020 May 12;21(10):3405. doi: 10.3390/ijms21103405. Int J Mol Sci. 2020. PMID: 32408480 Free PMC article. Review.
-
Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress.Antioxid Redox Signal. 2014 Jan 10;20(2):308-24. doi: 10.1089/ars.2012.4609. Epub 2012 Jul 13. Antioxid Redox Signal. 2014. PMID: 22657349 Free PMC article. Review.
-
NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease.Br J Pharmacol. 2017 Jun;174(12):1733-1749. doi: 10.1111/bph.13425. Epub 2016 Feb 26. Br J Pharmacol. 2017. PMID: 26750203 Free PMC article. Review.
-
Dual Character of Reactive Oxygen, Nitrogen, and Halogen Species: Endogenous Sources, Interconversions and Neutralization.Biochemistry (Mosc). 2020 Jan;85(Suppl 1):S56-S78. doi: 10.1134/S0006297920140047. Biochemistry (Mosc). 2020. PMID: 32087054 Review.
-
Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species.Biochim Biophys Acta. 2010 Jun-Jul;1797(6-7):897-906. doi: 10.1016/j.bbabio.2010.01.032. Epub 2010 Feb 1. Biochim Biophys Acta. 2010. PMID: 20122895 Review.
Cited by
-
Resolvin D1 promotes corneal epithelial wound healing and restoration of mechanical sensation in diabetic mice.Mol Vis. 2018 Apr 1;24:274-285. eCollection 2018. Mol Vis. 2018. PMID: 29643724 Free PMC article.
-
Oxidative Stress, Kinase Activation, and Inflammatory Pathways Involved in Effects on Smooth Muscle Cells During Pulmonary Artery Hypertension Under Hypobaric Hypoxia Exposure.Front Physiol. 2021 Aug 9;12:690341. doi: 10.3389/fphys.2021.690341. eCollection 2021. Front Physiol. 2021. PMID: 34434114 Free PMC article. Review.
-
Effects of ROS pathway inhibitors and NADH and FADH2 linked substrates on mitochondrial bioenergetics and ROS emission in the heart and kidney cortex and outer medulla.Arch Biochem Biophys. 2023 Aug;744:109690. doi: 10.1016/j.abb.2023.109690. Epub 2023 Jul 8. Arch Biochem Biophys. 2023. PMID: 37429534 Free PMC article.
-
BGP-15 Protects against Oxidative Stress- or Lipopolysaccharide-Induced Mitochondrial Destabilization and Reduces Mitochondrial Production of Reactive Oxygen Species.PLoS One. 2017 Jan 3;12(1):e0169372. doi: 10.1371/journal.pone.0169372. eCollection 2017. PLoS One. 2017. PMID: 28046125 Free PMC article.
-
Long-Term Effects of Aircraft Noise Exposure on Vascular Oxidative Stress, Endothelial Function and Blood Pressure: No Evidence for Adaptation or Tolerance Development.Front Mol Biosci. 2022 Jan 31;8:814921. doi: 10.3389/fmolb.2021.814921. eCollection 2021. Front Mol Biosci. 2022. PMID: 35174211 Free PMC article.
References
-
- Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM et al (2012). Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: potential implications for ROS signalling. Biochem J 441: 719–730. - PubMed
-
- Akizuki S, Yoshida S, Chambers DE, Eddy LJ, Parmley LF, Yellon DM et al (1985). Infarct size limitation by the xanthine oxidase inhibitor, allopurinol, in closed‐chest dogs with small infarcts. Cardiovasc Res 19: 686–692. - PubMed
-
- Almasalmeh A, Krenc D, Wu B, Beitz E (2014). Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J 281: 647–656. - PubMed
-
- Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015). The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172: 1587–1606. - PMC - PubMed
-
- Andrukhiv A, Costa AD, West IC, Garlid KD (2006). Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol 291: H2067–H2074. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
